
Abstract
First discovered in 1906, Alzheimer’s disease has become one of the most prevalent yet complex neurodegenerative diseases, impacting millions worldwide. Over a century of scientific research has yielded substantial progress in the understanding of this disease, with numerous proposed causations, implications, and medications. However, despite continuous clinical advancements, many issues concerning the treatment and medication of Alzheimer’s, such as economic drawbacks and severe adverse effects, including amyloid-related imaging abnormalities and cardiovascular complications, still persist; the multifaceted nature of this disease stymies rapid progress. This literature review bridges Guillain-Barré Syndrome (an acute autoimmune disease), Huntington’s disease, and Parkinson’s Disease, exploring their existing and developing therapies that have potential crossover application for Alzheimer’s disease, aiming to bring forth an innovative and comprehensive approach to examining Alzheimer’s treatment. Due to notable differences in pathophysiology and etiology of these diseases, they are not widely compared in tandem in current research. Nevertheless, emerging evidence attests to nuanced similarities in pathogenic pathways and interrelated causal nexus. This study attempts to expand the scope and perspective beyond current paradigms, utilizing underlying mechanisms of neurodegenerative and neurological autoimmune diseases as a basis for evaluating potential therapies and inventive crossovers.
Key Terms
1. Central Nervous System (CNS) – The complex of nerve tissues comprising the brain and spinal cord that controls the activities of the body
2. Peripheral Nervous System – The network of nerves located outside the brain and spinal cord that transmits signals between the CNS and the rest of the body
3. Ubiquitin–proteasome System (UPS) – Primary cellular pathway degrading unwanted or malfunctioning proteins
3. Amyloid-β (Aβ) plaques – Protein aggregates in Alzheimer’s, disrupting neural function
4. Tau tangles – Abnormal aggregates of hyperphosphorylated tau proteins, a key feature in Alzheimer’s
5. Blood-brain barrier (BBB) – A selective barrier limiting drug delivery to the brain
6. Deep brain stimulation (DBS) – Electrical modulation of brain circuits via implants
7. Antisense oligonucleotides (ASOs) – Synthetic RNA/DNA strands that block harmful protein production
8. Therapeutic plasma exchange (TPE) – Blood filtration to remove pathogenic antibodies
Methodology
Identification
A comprehensive search strategy was employed to examine academic sources published up to 2025. The review focused on a broad scope encompassing basic molecular mechanisms (pre-clinical) through to clinical trial stages (I, II, and III) for various neurodegenerative diseases, with a central focus on Alzheimer’s Disease. Primary sources included journals and databases from leading institutions such as the National Institutes of Health (NIH), International Journal of Molecular Sciences, National Institute on Aging, and Frontiers in Human Neuroscience.
Screening
Records retrieved from the initial search were screened based on their titles and abstracts. This step excluded a significant number of records that were outside the defined scope (e.g., unrelated disease fields, non-neurological focus), were not primary research or authoritative reviews, or were published in non-academic sources.
Eligibility
The full text of the remaining articles was critically assessed for eligibility. The inclusion criteria were:
Population/Subject: Research pertaining to human patients, animal models, or cellular models of neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s, Huntington’s, Guillain-Barré Syndrome).
Intervention/Exposure: Studies on disease mechanisms (e.g., protein aggregation, neuroinflammation, mitochondrial dysfunction), diagnostics (e.g., biomarkers, imaging), and therapeutics (e.g., monoclonal antibodies, deep brain stimulation, plasma exchange, antisense oligonucleotides).
Outcome: Measurable outcomes related to pathological understanding, biomarker levels, cognitive/functional improvement, or safety profiles.
Study Type: Original research articles, systematic reviews, clinical trial reports, and authoritative guidelines from major health organizations.
Included
A total of 150 sources met all eligibility criteria and were included in the final qualitative synthesis, forming the cited bibliography for the literature review. After numerous rounds of screening, 11 sources met the criteria for discussion of clinical trials. These sources provide the foundational evidence for discussing disease pathways, diagnostic approaches, and the current landscape of therapeutic development.
Disease Background
With an ever-expanding catalogue of known neurological and neurodegenerative diseases, perceiving similarities and distinctions between them could be the first step in tackling unsolved enigmas and advancing novel therapeutic approaches. Beyond concentrating focus on Alzheimer’s disease in examining disease mechanisms, pathophysiology, and existing treatments, analysis of Guillain-Barré Syndrome, Huntington’s Disease, and Parkinson’s Disease is also compared and discussed below.
Alzheimer’s disease: UPS dysfunction and Neuroinflammation
Alzheimer’s, contributing to 70% of all dementia cases, has impacted over 55 million people across the globe1. Alzheimer’s disease is a severely destructive disorder that leads to physical and mental disintegration, impacting the welfare of not only diagnosed patients but also severing social ties with families and communities. With no known cure thus far, Alzheimer’s remains the 7th leading cause of death globally, claiming more than a hundred million lives annually2.
Alzheimer’s disease is postulated to potentially involve both autoimmune and inflammatory mechanisms, with protein aggregation being a hallmark of this disease3. As a neurodegenerative disorder that manifests through a progressive loss of episodic memory and cognitive function, Alzheimer’s subsequently leads to language impairment, emotional and psychiatric dysregulation. Neuropathological degradation progresses over 3-8 years, depending on the time of diagnosis and life expectancy of each patient4. Most existing treatments have evolved around slowing down disease progression, strengthening neurons, and removing potential obstructions around transmission pathways5.
Though with an unclear prognosis, an internal or external trigger is associated with the abnormal buildup of proteins, disrupting the regular functioning of neurons by the toxicity of the proteins, and also becoming a physical barrier to transmissions6. Diverse causes of Alzheimer’s have also been explored with various findings. Studies have examined the potential role of the ubiquitin-proteasome degradation system (UPS) in neurodegeneration. The UPS is a complex system of proteins with a major function of intracellular protein regulation7. In the normal UPS degradation pathway, irregular protein formation is detected and tagged by a regulatory protein, ubiquitin. The tagged protein is recognised by a proteasome complex, which contains catalytic enzymes that cleave polypeptide chains into amino acids, allowing the recycling of faulty or unused proteins. Research by Hsieh et al. reveals that Alzheimer’s could be interrelated with UPS impairment, as neurons with lower UPS activity displayed greater vulnerable to protein aggregation and accumulation8. This finding proposes a pathway of UPS malfunction leading to cellular inability to degrade unwanted proteins promptly, causing further buildups into insoluble plaques and fibrils.
Another subsequent cause explored is neuroinflammation in causing protein accumulation9. Glial cells, which form the primary structural composition of the myelin sheath, are activated by neuroinflammation, triggering inflammatory responses such as the production of pro-inflammatory proteins10. These proteins, known as cytokines, can lead to neuron degradation and cell death, even aggravating other disease pathways such as atherosclerotic plaque build-up in arteries11. Beyond medicinal approaches, other therapeutic strategies have been explored, targeting newly proposed pathological pathways and mechanisms12.
Guillain-Barré Syndrome: Maladaptive Plasticity and Perfusion Abnormality
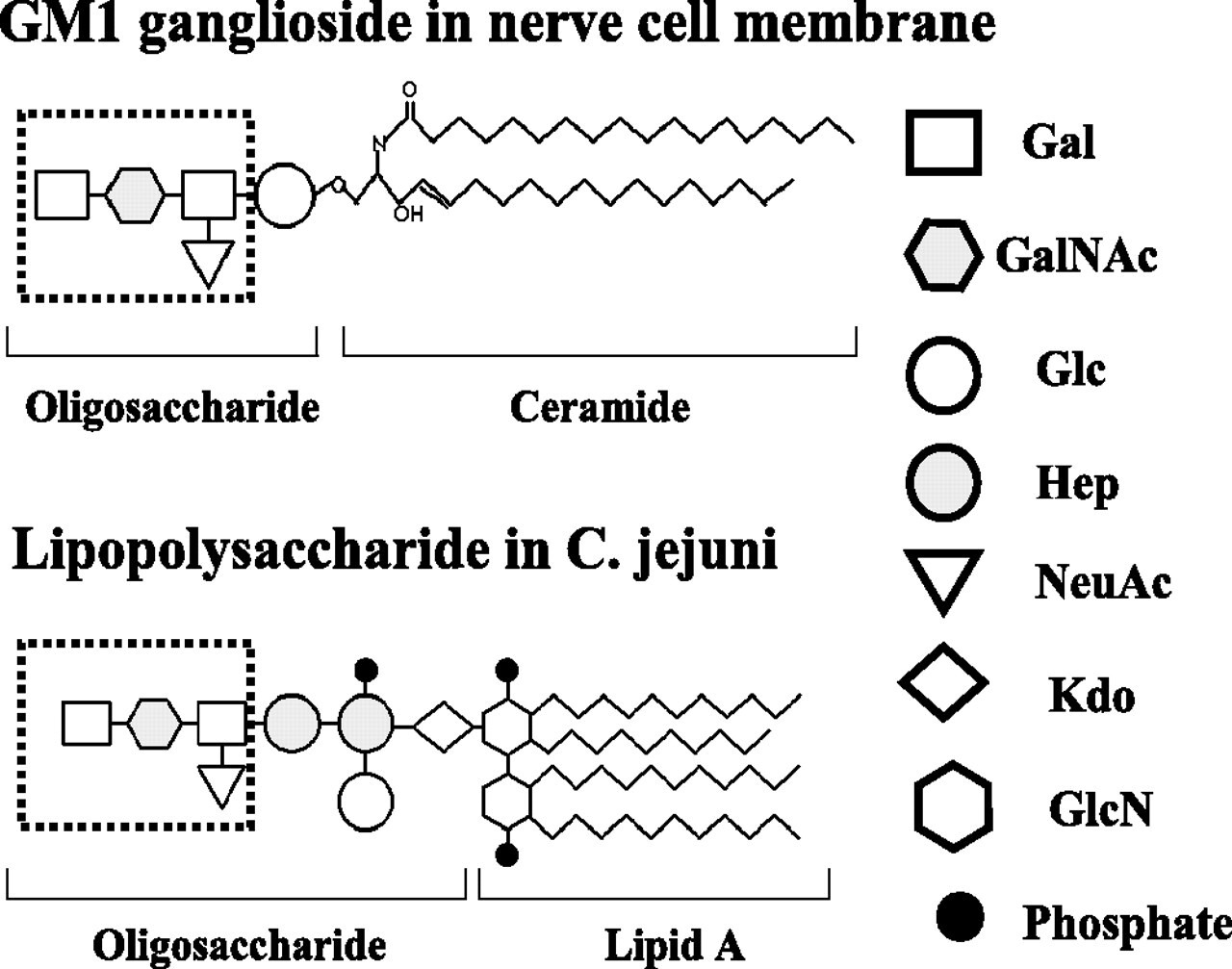
Guillain-Barré Syndrome is a severe autoimmune disorder that leads to paralysis, dysphagia, and damage to the autonomic nervous system13. Despite its seeming dissimilarity to neurodegenerative disorders, the pathophysiology of Guillain-Barré syndrome is closely linked to neuron transmission, as it severely impacts the peripheral nervous system14, and is caused by the infection of specific viruses such as the Campylobacter or Cytomegalovirus. Upon infection, a digestive tract or respiratory infection triggers an immune response, instigating B-cells to produce antibodies that neutralise viruses and prevent further pathogenic infection15. However, in a phenomenon termed “molecular mimicry”, the resemblance between lipopolysaccharides on viral cell walls and ganglioside receptors located on the axon or myelin sheath induces an autoimmune response, the antibodies attacking gangliosides such as GM1, GD1a, and GQ1 instead16 (Fig. 1). The damage done to the protective myelin layer causes interference with axon body transmission, slowing electrical impulses and potentially causing a signalling termination17. The peripheral neuroinflammation and nerve damage to neuromuscular junctions contribute to varying physiological symptoms, ranging from acute motor axonal neuropathy to acute pandysautonomia18.
Given the bidirectional interdependence between the peripheral nervous system and the central nervous system (CNS), there may be pathological implications of peripheral neuropathy in disrupting CNS processes. Maladaptive plasticity caused by peripheral nerve damage leads to a series of dysfunction and reorganisation of transmission networks, resulting in extreme hypersensitivity in spinal cord neurons20. With intensified sensory signalling, pro-inflammatory cytokines (such as Interleukin-1 beta) are triggered to be released in the spinal cord, not only causing immune dysregulation, but also impacting brain regions such as the microglia by instigating the overproduction of reactive oxygen species21. These ramifications could trigger further inflammation and create a negative feedback loop, or induce changes in microglial morphology, potentially predisposing other chronic illnesses or increasing the severity of neurodegeneration22.
Another proposed link between Guillain-Barré Syndrome and CNS impairment is perfusion abnormalities. Due to autonomic dysfunction, Guillain-Barré Syndrome could result in rare causes of Posterior Reversible Encephalopathy Syndrome, impairing regulation and sometimes hypoperfusion23. Damage to the blood-brain barrier (BBB) could be the result of a “leaky brain”. Potential harmful substances like plasma proteins and inflammatory molecules may thus be able to bypass barriers and enter the brain, leading to a cascade of events such as vasogenic edema that could exacerbate chronic neurodegeneration24. However, due to prominent differences between Guillain-Barré Syndrome, an acute autoimmune disorder, and neurodegenerative diseases, further studies and investigations are required to establish concrete causality and pertinence.
Huntington’s Disease: Glutamate excitotoxicity and Synaptic dysfunction
Huntington’s disease is a hereditary disease leading to loss of conscious motor control and emotional regulation abilities25. In Huntington’s disease, a genetic mutation on the HTT gene causes an abnormal number of repeats of a certain codon CAG when producing polypeptide chains. The surplus of CAG codes for a lengthened chain of glutamine amino acids alters the final quaternary conformation of the produced protein known as huntingtin (Htt), the changes in bonding interactions causing mutated huntingtin (mHtt) proteins to form clusters and aggregate, accumulating around neurons26. It is hypothesised that the protein aggregates cause blockages of nuclear pores within the nucleus located in the soma, restricting movements of chemicals and energy compounds, leading to decreased transmission efficiency and neuronal cell death27. Similarly, mHtt protein aggregates also cause heightened excitotoxicity by impairing glial cell function. mHtt protein induces genetic and morphological changes in astrocytes by sequestering transcription factors and subsequently gene expression28, impacting numerous processes such as glutamate reabsorption across synapses. Glutamate is proposed to bind and activate NMDA receptors, causing calcium ion influx into neurons, inducing oxidative stress, and disrupting action potential transmission29. As an excitatory neurotransmitter, glutamate similarly plays a crucial role in the pathophysiology of other neurological diseases, including Alzheimer’s disease30. Excitotoxicity caused by excessive NMDA activity is a proposed pathological mechanism of Alzheimer’s, with excess Ca2+ indirectly hyperphosphorylating tau proteins – a key hallmark of Alzheimer’s neurodegeneration. Calcium ions stimulate the formation of numerous biomolecules, such as lipid molecules, PGE2, that contribute to the phosphorylation of tau proteins, and later catalyze aggregation31. Beyond this, due to several analogous symptoms and underlying etiological similarities, Huntington’s and Alzheimer’s have further similarities in disease pathway and mechanisms, summarised in Table 1 below.
| Major disease pathway | Analogous mechanism |
| Protein aggregation | Misfolding and aggregation of proteins32 |
| Impairment of the ubiquitin proteasome system (UPS) through proteasome catalytic site binding and capturing free ubiquitin molecules33 | |
| Sequestration of Beclin-1 causes impaired autophagy regulation by influencing recognition between the autophagosome and ubiquitin-tagged cargo or autophagosome-lysosome binding34‘35, 26/11/2025 17:07:00 | |
| Phosphorylation and Palmitoylation of diseased proteins perpetuating aggregation36 | |
| Synaptic dysfunction | Increased extrasynaptic NMDA receptors and phosphorylation as a potential consequence of increased Ca2+ influx37 |
| Altering synaptic plasticity by reducing brain-derived neurotrophic factor levels, reducing transportation efficiency, and impeding long-term potential (LTP) and long-term depression (LTD) processes38 | |
| Mitochondrial dysfunction and defects in substance trafficking39 | |
| Release of neurotoxic metabolites from the kynurenine metabolism pathway causes damage to the central nervous system, including oxidative stress and neuroinflammation40 |
Though with evident convergent pathways, Huntington’s and Alzheimer’s do possess many disparate variations, like a monogenic and hereditary cause compared to a polyphasic cause for Huntington’s and Alzheimer’s, respectively. Similarly, Huntington primarily impacts selective motor mechanisms, while Alzheimer’s and other CNS disorders entail cognitive and memory disruption. Nevertheless, these proposed similarities may be worth exploring, giving rise to integration possibilities in diagnosis and therapeutic approaches.
Parkinson’s Disease: Genetic predisposition and Mitochondrial failure
Parkinson’s Disease is the second most common dementia after Alzheimer’s, and is similarly associated with toxic protein aggregates in causing motor control dysregulation and cognitive decline42. Varied causes have been proposed for Parkinson’s Disease, mainly including genetic bases and environmental stimuli such as exposure to neurotoxins. Exposure to chemical muta-gens like pesticides and head trauma, cause epigenetic alterations, trigger neuroinflammation, and catalyze protein aggregation43. However, variability in genetics can also play a role in influencing physical responses to environmental stimuli, causing differences in disease progression even among patients in similar environs44. The significance of epigenetics is especially noted in numerous studies, highlighting the interrelatedness between environmental neurotoxic triggers and their consequences in impacting the epigenome, with generational repercussions through transgenerational epigenetic inheritance. The plausibility of multiplex environmental and genetic origins applies to numerous neurological and neurodegenerative diseases45.
Exploring potential genetic influences in increasing abnormal protein accumulation, a comprehensive review by Tran et. al. convened 67 studies and concluded that numerous analogous phenotypes were shared among genetically heterogeneous patients, highlighting the potential of genetic influence in inducing disease pathways46. The study emphasised links between gene expression patterns and both α-synuclein proteins and tau fibrils, including elevated expression of the SNCA gene alongside other responses such as proteosome impairment47.
Conclusive causes of Parkinson’s are still being explored and verified, but numerous studies have pointed towards the primary basis of dopaminergic neuronal dysfunction, with obstruction in the basal ganglia, and hence leading to motor control impairment and disrupted mood regulation48. A key feature associated with Parkinson’s is the aggregation of Lewy bodies. Composed mainly of alpha-synuclein protein (α-synuclein) and ubiquitin, these abnormal protein clumps cause significant obstruction in the neurotransmission pathway49. Although the body system contains numerous mechanisms for degrading and removing mutated proteins or waste products, α-synuclein proteins are speculated to be able to resist cellular degradation by affecting autophagosome vesicles that transport particles for lysosomal degradation50. The protein buildup mainly occurs in the subcortical regions, as indicated in Figure 2. By altering neuronal structure and processes in the substantia nigra, dopamine transmission to the cortex is severely hindered, resulting in conflicting signalling that impedes cognition and motor movements51.

Another proposed pathology pathway is mitochondrial failure. The causation relationship between mitochondria, astrocytes, and neurons was termed as the “Mitochondrial Triad Hy- pothesis” in a recent 2025 study by Walecha and Luthra53. A bidirectional relationship between neuronal cells and astrocytes is the basis of the metabolic functioning of neuronal networks, with exchange of lactate as an energy source, glutamate buffering, and supply of healthy mitochondria54. Astrocytic mitochondria, especially, play a crucial role in energy production, regulating calcium homeostasis and activating controlled cell death55. Following the pathological process of Parkinson’s, astrocytes sometimes undergo conformational changes that result in functional deterioration. In response, cytokines and reactive oxidative species are released, causing heightened neuronal stress. This could then cause increased α-synuclein accumulation, inflammation, and even cell death. This feedback loop of consistent disruption may cause severe inflammation, exacerbating neurodegeneration by both increasing protein aggregation and causing chronic inflammation56. The astrocyte dysfunctional pathways are depicted in Figure 3 below. Furthermore, other studies have pointed towards further interrelationship between astrocytic degradation and neurodegeneration of Parkinson’s, with potential relevance in other neurodegenerative diseases as well57.

Types and pathophysiology of neurodegenerative and neurological conditions may vary significantly, with the examples of Guillain-Barré Syndrome, Huntington’s disease, and Parkinson’s Disease illustrating different progression pathways and hypothesised causes. The understanding of these varied diseases could provide insights into therapeutic approaches, with both similarities and distinctions enabling new perspectives that may lead to novel treatments.
Pathophysiology of Alzheimer’s disease
The cause of Alzheimer’s disease remains elusive, with various hypotheses linking ageing, lifestyle, and genetic influences catalysing the development of this neurodegenerative disease59. Studies have identified a specific gene variant, APOE4, to be strongly associated with Alzheimer’s development risk, associated with numerous pathological pathways. Carriers of two copies of the APOE4 gene were predicted to have a 60% higher likelihood of developing Alzheimer’s in later stages60, linked with increased accumulation of β-amyloid and neuroinflammation61. Similarly, environmental risk factors have also been proposed. A meta-analysis examining correlated risks of different exposure factors with heightened probability of Alzheimer’s development proposed that increased exposure to specific fine particulate matter PM 2.5, PM 10, and ozone was associated with an increase in Alzheimer’s risk factor62.
Though with differing proposed factors predisposing Alzheimer’s disease, a hallmark of this neurodegenerative disease is abnormal protein buildups, specifically β-amyloid and tau proteins, forming amyloid plaques and neurofibrillary. β-amyloid (Aβ) is derived from the breakdown of amyloid precursor proteins (APP). Seen from Figure 4, the normal cleavage pathway (non-amyloidogenic) involves cleavage of APP into subunits of soluble peptide sAPPα and APP intracellular domain (AICD) by α-secretase enzyme. The synthesis of sAPPα and AICD fragments allows neurotrophic and neuroprotective functionality, allowing optimal chemical synaptic transmission, as well as regulation of neuronal genes63.

In the amyloidogenic pathway, the formation of Aβ is caused by sequential cleavage: first by β-secretase (producing C99), followed by γ-secretase cleavage, which generates both Aβ peptide and AICD.65. This results in the accumulation of Aβ oligomers and amyloid plaques that inhibit regular chemical transportation and accelerate neuronal death66. Aβ plaques also cause chemical dysfunction due to interactions with protein receptors, such as the alpha-7 nicotinic receptor (α7 nAChR), which is implicated in long-term memory, influencing the nervous system and hence leading to triggers of inflammatory responses that cause neuronal death. Aβ oligomers are also hypothesised to trigger the formation of a subsequent protein. When Aβ is formed by the cleaving of APP, it can activate specific enzymes such as CDK-5 and GSK-3β67, which play important roles in the phosphorylation of tau protein.
Tau is a protein isoform typically found around the soma and dendrites68, maintaining the structural stability of the neuron cytoskeleton. In a diseased brain, a process of hyperphosphorylation occurs, where excessive phosphorylation attaches phosphate groups onto the neuron microtubule. This, in turn, leads to depolarisation, where tau proteins detach from microtubules. Without tau protein binding, the microtubule becomes structurally unstable and causes disruptions to neuronal signalling69. Additionally, due to the altered chemical composition of phosphorylated tau proteins, they can bind and form tau oligomers, known as neurofibrillary tangles (NFTs). NFTs not only cause physical blockages but can also obstruct the neurotransmission pathway, inhibiting synaptic energy production that leads to cell death70.
Hence, to combat protein aggregation, pharmaceuticals such as lecanemab, donepezil, and galantamine have been developed to target protein clearance and inhibit enzymes to increase neurotransmission across healthy neurons. Nevertheless, many limitations remain with sole reliance on pharmaceutical interventions. This is attributed to the complexity of the nervous system and the human brain, with impediments such as the blood-brain barrier severely hindering drug delivery.
Diagnosis and early detection of Alzheimer’s neurodegeneration
Given the polyphasic and progressive nature of Alzheimer’s disease, early detection is crucial for preventative treatments. With amyloid and tau proteins established as conspicuous indications of neurodegeneration, diagnostic methods, including PET and CSF tests, mainly revolved around the detection of amyloid plaques and tau fibrils. Position Emission Tomography (PET) tests involve the injection of radioactive ligands of amyloid or tau protein into the patient’s bloodstream, allowing it to travel through the vascular system before reaching the cerebral vasculature71. Upon reaching the brain endothelial cells, the radioligand binds to specific (LRP1) receptor proteins, allowing internalization across the blood-brain barrier in a process known as receptor-mediated transcytosis72. This process is also explored for potential in aiding the transportation of therapeutics and nanoparticles into the brain73. Due to structural compatibility between the ligands and their targeted protein, the radioactive ligands can bind to amyloid plaques or tau protein aggregates if they are present in the brain, enabling radioactive detection via the PET scanner. PET examinations allow accurate detection of specific protein presence in the brain, and are also non-invasive and low-risk procedures. However, its high cost at $3000 per scan, which causes significant burdens with consistent testing for disease monitoring74.
Another common diagnosis technology is the Cerebrospinal Fluid test (CSF), which involves a lumbar puncture to collect cerebrospinal fluid that can be used for biomarker analysis. Common cerebrospinal fluid biomarkers for Alzheimer’s disease include amyloid variant Aβ1-42 and tau phosphorylated at T181 (p-tau181)75. Emerging research has also identified Visinin-like protein-1 (VILIP-1), a calcium-binding protein, as a potential biomarker for neuronal damage, with it being released to the cerebrospinal fluid upon neuronal death76. With rapid progress in the identification of protein variants as biomarkers, alternative non-invasive blood tests have also been explored. A test by Grande et al. evaluated the efficacy of six blood markers in predicting Alzheimer’s, with findings revealing that heightened baseline levels of p-tau217, p-tau181, neurofilament light chain, and glial fibrillary acidic protein (GFAP) were associated with increased risk of pathology development, especially noting the efficacy of combined p-tau217 and GFAP in Alzheimer’s specific dementia diagnosis77.
Limitations of existing therapies
Blood-Brain Barrier
The Blood-Brain Barrier (BBB) is a major impediment to many neurological and neurodegenerative disease treatments, limiting direct access to the brain tissue. However, BBB’s vital role necessitates cautious approaches, as disruptions lead to severe and oftentimes irreversible damage78.
Mainly composed of endothelial cells, basal lamina, and astrocytes, the BBB prevents perturbation by posing physical lines of defence between neurons and blood vessels. Tight junctions between endothelial cells control diffusion through the membrane, with the basal lamina and macrophages similarly contributing to the secondary filtering of chemical substances. Astrocytes also play a crucial role in maintaining the structural integrity of the BBB by surrounding the blood vessels with astrocyte end-feet, also influencing endothelial tight junctions by transmitting chemical signals79. The BBB plays a crucial role in separating cellular environments, maintaining homeostasis, and preventing toxins from entering into the brain. However, this similarly gives rise to major impediments in drug delivery, as the BBB only permits substances with molecular masses of less than 400-500 Dalton to pass through, especially restricting crossing of hydrophilic or charged molecules.80. Given this, numerous studies and trials have investigated methods to bypass this barrier.
One major focus is the application of nanoparticles, specifically lipid-based nanomaterials, as drug delivery capsules. Lipid-based nanomaterials are synthetic particles with hydrophilic or amphipathic properties due to their non-polar hydrocarbon chains. Utilizing this property, lipids can form micelles or bilayers that separate intracellular and extracellular environments by introducing two distinct hydrophobic and hydrophilic regions. As a novel drug delivery system, lipid-based nanomaterial allows clear separation from the exterior environment, effectively maintaining optimal conditions such as pH, osmotic stability, and concentration81. Given lipid nanoparticles’ unique properties, they can pass through the BBB using the mechanisms including carrier-mediated transcytosis, receptor-mediated transcytosis, and absorptive-mediated transcytosis82.
A subsequent method targeting BBB opening is focused ultrasound. Focused ultrasound (FUS) is a non-invasive technology allowing a temporary opening of the BBB by using low-intensity soundwaves to manipulate intravenously injected microbubbles (MB)83. Microscopic gas-filled bubbles with a lipid or protein sphere are injected into the bloodstream, and as MRI-guided ultrasound waves penetrate soft tissue and bone, they target specific locations with high accuracy. The focused ultrasound sonication causes cavitation effects, where microbubbles oscillate and contract, the movement generating pressure against endothelial cell walls, temporarily disrupting tight junctions and thus increasing the permeability of the BBB84 (Fig. 5).

Clinical trials have been ongoing to test the efficacy of this technology in a wide range of applications, aiding drug delivery and treatment for neurodegenerative diseases. With the prominence of BBB in brain disorder treatments, numerous other focus areas have also been proposed, including utilizing existing protein transporters such as LRP186, or invasive methods of cerebrospinal fluid and intracranial delivery87.
Monoclonal antibodies: Lecanemab
Lecanemab was approved by the FDA in July 2023 for treatment against amyloid protein aggregation in mild-Alzheimer’s disease. Lecanemab is a monoclonal antibody synthesised by altering a murine-derived antibody88, which recognises and selectively binds to Aβ aggregates and oligomers, specifically by recognising amino acids 1–16 and 21–29 of Aβ peptide89. Lecanemab facilitates the clearance of Aβ proteins by various proposed mechanisms, including destabilisation of fibrils to break down protein aggregates into smaller fragments, decreasing the obstruction and entanglement caused by protein fibrils90. Another mechanism proposed includes microglial phagocytosis, where antibodies interact with microglial receptors, initiating phagocytic mechanisms for protein clearing91.
Lecanemab has been shown to benefit protein clearance and enhance cognitive decline by consequently reducing tau phosphorylation and neuroinflammation. A wide-scale trial demonstrated a 26% slowing in cognitive decline by reduction of amyloid plaques, but was limited to efficacy in early-Alzheimer’s patients, unable to support the treatment of patients with mild to severe Alzheimer’s disease92. With the majority of current Alzheimer’s patients being of elderly age due to late detection of the disease, many with more severe neurodegeneration are unable to receive lecanemab treatment. Furthermore, given the mechanism of action of lecanemab when removing fibrils encapsulated around blood vessels, it could, in turn, damage neuronal blood vessels, leading to haemorrhage. A phase III trial conducted reported results of numerous cases of acute intracerebral haemorrhage after lecanemab infusion treatments, causing two patient deaths that were attributed to the adverse effects of the drug93. This, alongside several limitations of efficacy and cost, has limited the applicability of lecanemab as a long-term treatment for Alzheimer’s disease.
Apart from Lecanemab, numerous other therapeutic drugs have been tested for Alzheimer’s, including recently approved Donanemab, and experimental monoclonal antibodies Remternetug and Trontinemab, both reaching Phase III clinical trial stages94. Conversely, therapeutics such as Solanezumab were also unable to achieve statistically significant impacts to inhibit or clear Aβ plaques, and clinical trials were terminated early due to the occurrence of extreme adverse effects of lymphocytopenia and even skin cancer95. Major monoclonal antibodies applied or tested for Alzheimer’s disease are summarised in Table 2 below.
| Monoclonal antibody | Clinical status | Mechanism of action & efficacy | Adverse effects | Cost & Availability |
| Lecanemab (Leqembi®) | Approved (2023) | Targeting soluble Aβ protofibrils, aiding protein clearance upon neutralisation Showed 25% higher on ADCOMS* compared to placebo | – Amyloid-Related Imaging Abnormality – Effusion (ARIA-E) (9.9% patients out of 609 subjects with early disease progression)96- Mild to moderate infusion reactions – Autoimmune encephalitis (92 compared to 15 in the placebo group) | Yearly cost: $26,500 Appropriate for patients with early Alzheimer’s, excluding those on anticoagulants or who have blood-clotting disorders97 |
| Donanemab (Kisunla®) | Approved (2024) | Binds to modified Aβ plaques, facilitating removal via microglial -mediated phagocytosis | – ARIA-E (23.9% patients out of 71 subjects)98 – Sepsis-associated encephalopathy (18.3%) | Yearly cost: $32,000 Appropriate for patients with mild cognitive impairment, including those with prior unstable illnesses or who do not meet MRI screening criteria99 |
| Aducanumab | Phase III discontinued in 2024 | Binds to aggregated Aβ, including both oligomers and fibrils, promoting clearance | – ARIA-E (35% out of 537 patients receiving high dose) – Localized superficial siderosis (13%) | Discontinued due to lack of efficacy and safety concerns. |
| Remternetug | Phase III | Remove aggregated Aβ via microglial -mediated phagocytosis | – ARIA (41% out of 24 patients)100 | Trials in progress |
| Trontinemab | Phase III | Binding to fibrillar Aβ plaques, while coupled with a transferrin receptor shuttle module for removal101 | – Phase 1a/2b trial had no ARIA-E adverse effects, and 1 ARIA-H incident102 | Trials in progress |
*ADCOMS = Alzheimer’s disease Composite Score
Many therapeutics in development for Alzheimer’s disease treatment, though having the potential to aid protein aggregate clearance, are marred by severe adverse effects, most dominantly Amyloid-Related Imaging Abnormality – Edema/Effusion (ARIA-E). Furthermore, with the approved and released monoclonal antibody drugs, high prices remain a significant issue that limits wide-range accessibility for many patients. The efficacy relative to its risk and cost urges further development of diverse therapeutic approaches.
Therapeutic Approaches
Current major therapies of Alzheimer’s revolve around pharmaceutical interventions with monoclonal antibodies, but due to limited efficacy for full clearance of protein aggregates and adverse effects, coupled with high cost and limited availability, make it a somewhat inaccessible treatment option for many patients. Although the pathophysiology and disease pathways of the discussed neurological and neurodegenerative diseases (Guillain-Barré syndrome, Huntington’s Disease, and Parkinson’s Disease) are distinct, and thus limit direct extrapolation and translation, many of their therapeutic approaches and strategies have great potential for integration or prompting new treatment designs. The therapies to be discussed include Deep Brain Stimulation and focused ultrasound, therapeutic plasma exchange, antisense oligonucleotide, and gene therapies.
Neurosurgical procedure: Deep Brain Stimulation & Focused Ultrasound
Deep Brain Stimulation (DBS) is a minimally invasive treatment commonly applied to the treatment of neurological movement disorders, such as the aforementioned Guillain-Barré syndrome and Huntington’s disease, the efficacy of each evinced by case reports from J. Theuriet et al.103 and Wojtecki et al.104. The treatment was also approved by the FDA as a procedure for Parkinson’s Disease early in 2002 (NIH), enabling over 5500 annual admissions of DBS treatment105.
This treatment involves the implantation of neurostimulators and electrodes into targeted regions of the brain, which direct electrical impulses deep within brain cell nuclei106. Various mechanisms of DBS include depolarisation, inactivation, and inhibition of neuronal cells and electrical signals107. By modulating abnormal neurotransmission signals, DBS could alleviate symptoms and restore normal neuronal messaging108, reducing symptoms of neurodegenerative diseases caused by disrupted and irregular electron signalling. In Parkinson’s Disease, the major disease pathway is the impaired signalling and disrupted neuronal modulation, mainly in the basal ganglia-thalamocortical motor circuit (see Fig. 6). The disrupted neuronal signalling causes dopaminergic cell death, which in turn causes motor control dysfunction.
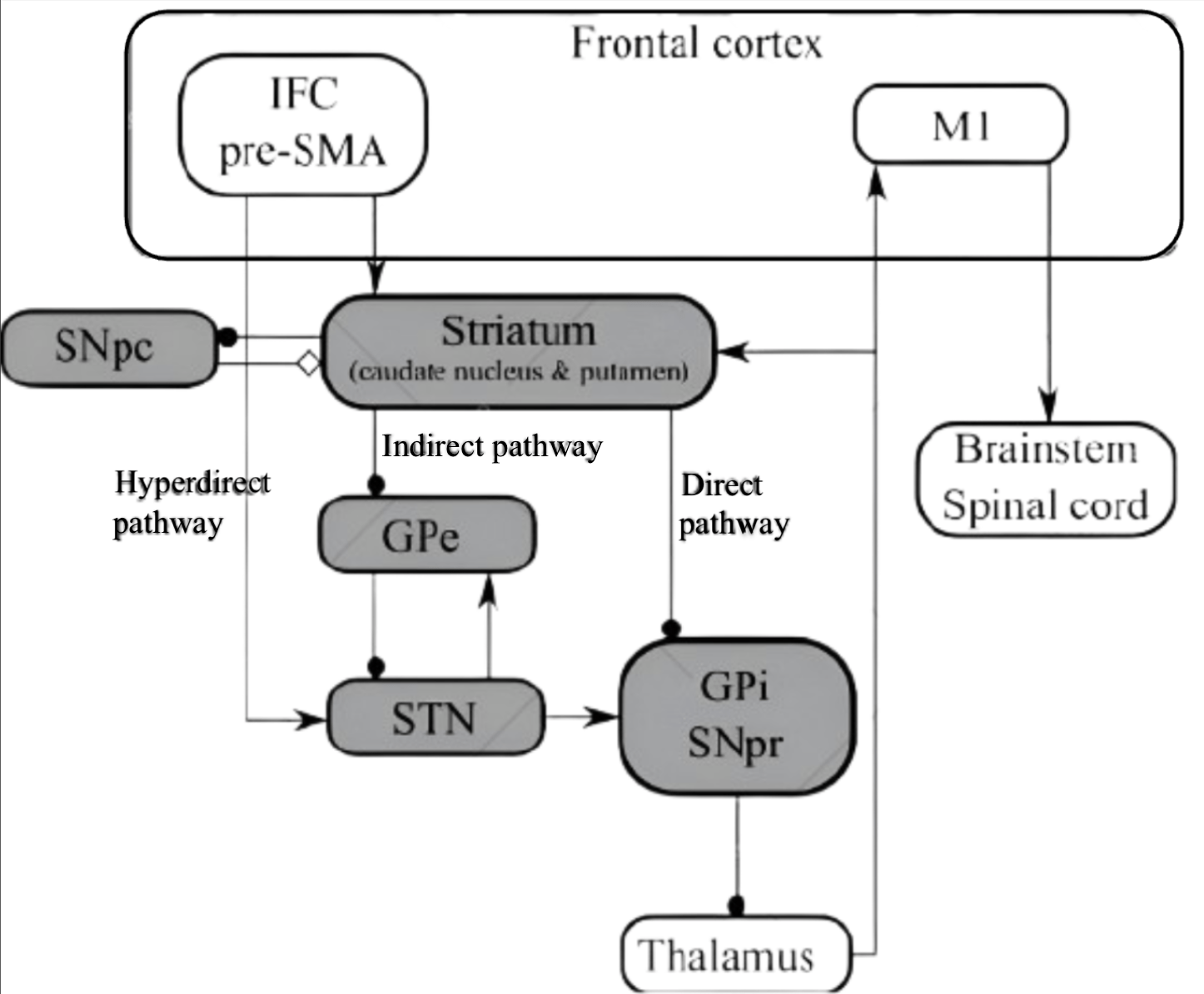
In Parkinson’s Disease, DBS mainly targets the subthalamic nucleus (STN) or Globus pallidus internus (GPi) in the basal ganglia region, transmitting high-frequency electrical waves that regulate neuronal electrical transmissions. This thus enables a degree of restoration of normal transmission in this circuit, enhancing cognition and facilitating motor control.
Alzheimer’s disease, on the other hand, though it does not include a sensorimotor circuit, is similarly associated with disruptions of neuron transmission due to physical blockage and chemical interference. Numerous studies have explored the application of DBS in Alzheimer’s disease, targeting major areas of the fornix (white matter connecting the hypothalamus to the hippocampus), nucleus basalis of Meynert, and the ventral striatum, all associated with memory processing and complex cognitive function110. In the earliest study in 2008, DBS was applied to target the fornix, with results showing increased electrical activity in the hippocampus, with the external electrical signals stimulating transmission111. In a later study by Laxton et al., 6 patients with mild Alzheimer’s received a DBS electrode implantation targeting the fornix region for 12 months, assessing the efficacy of the treatment based on cognitive function and measuring neuronal activity. The study concluded with observed cognitive improvements of some patients and deceleration of neurodegeneration, specifically the restoration of normal neuronal activity in the fornix upon DBS stimulation112. It was thus proposed that the potential mechanism of DBS in Alzheimer’s treatment targets key nodes in brain circuits, improving transmission connectivity and thus enhancing cognitive function.
Another postulated mechanism is reducing Aβ aggregates and tau fibrils. A mouse model study in 2010 applied DBS to the hippocampal slices, which revealed electrical signals to have prompted α-secretase activity while maintaining normal activity of β-secretase. Given the significance of both α-secretase and β-secretase (see Fig. 4) in the non-amyloidogenic and amyloidogenic pathways, respectively, stimulation of α-secretase promotes regular cleavage pathways to enable neurotrophic activity and neuroplasticity, without resulting in the formation of Aβ peptide. This suggests the capability of DBS in preventing Alzheimer’s pathology, with the potential of directly interfering with the primary pathological pathway of abnormal protein formation113.
However, evaluating the safety and risks of DBS treatment, a trial by Leoutsakos et al., including 42 Alzheimer’s patients, reported that more than half who received treatment experienced adverse effects, and 5 patients experienced more serious complications. Although a minimally invasive procedure, DBS treatment does contain risks of infection, hemorrhage, and stroke114. Another analysis examining DBS for Parkinson’s Disease found 23.3% of 1,079 patients who underwent DBS treatment passed away between 2 to 14 years after the procedure115. Although a causation relationship isn’t clearly delineated, it does raise the question of safety for this treatment. From the study, patients were carefully screened to match testing criteria due to surgical risks, medical history of intracranial haemorrhage, and stage of disease. Due to the potential risk associated with DBS, a limited number of patients are approved for treatment, with 1.6 % out of over 600 Parkinson’s patients suitable for surgical implantation for DBS116. Similarly, for Alzheimer’s, age and stage of disease remain significant restrictions for enabling wider application of DBS, as highlighted in a trial reporting adverse effects on patients under 65 years old117, hence highlighting the limited flexibility and adaptivity of DBS treatments.
Though trials have shown potential benefits of applying DBS to Alzheimer’s treatment, there are still significant limitations regarding safety and efficacy, requiring further investigation to clarify the mechanism of action and potentially integrate other technologies to minimise adverse effects.
A possible approach is to integrate focused ultrasound (FUS) therapy. FUS is currently explored in BBB opening technology to aid drug delivery for neurodegenerative disorders, through concentrated ultrasound radiation, manipulating microbubble oscillation. However, similar to DBS, FUS also has therapeutic applications in tremor suppression treatments, potentially also being integrated in Alzheimer’s treatments in aiding neuronal transmission118. Ultrasound therapies have been investigated to influence numerous mechanisms, including stimulating the production of pro-inflammatory cytokines and endogenous neurotrophins, proteins that are crucial in the regulation and development of the nervous system119. Application of targeted acoustic energy may activate microglia and astrocytes, enhancing protein clearance. A trial applying low-intensity pulsed ultrasound to mouse models with dementia noted significant improvement in cerebral blood flow, as well as increased expression of olig2-positive oligodendrocyte precursor cells (OPCs), which aid in the process of myelination and phagocytosis. Noticeably, the report noted a decrease in APP (amyloid precursor protein) expression and β-site amyloid precursor protein cleaving enzyme-1, which may subsequently lower APP cleavage into Aβ protein120. Furthermore, FUS may also present benefits in safety and accessibility. Unlike DBS, FUS is a non-invasive procedure that doesn’t involve surgical intervention. A study by Nicodemus et al. validated no adverse effects on all 10 dementia patients after applying low-intensity ultrasound neuromodulation121.
Thus, as a non-pharmaceutical therapy, DBS for Parkinson’s Disease is presented to have potential applications for Alzheimer’s, even with integration potential with existing technologies such as FUS already commonly implemented in Alzheimer’s disease.
Therapeutic plasma exchange: Albumin replacement
Therapeutic plasma exchange (TPE) is a treatment applied to autoimmune diseases that involves an extracorporeal intervention by removing and recirculating blood plasma from the patient. After extraction, plasma is separated via centrifugation from other components and filtered to remove toxic chemicals or components that are causing the autoimmune disease. The patient’s own filtered and processed plasma is then returned via infusion, or in some cases, plasma taken from healthy donors is then injected back to the patient. TPE is a common immunotherapy for Guillain-Barré Syndrome, as it supports the removal of B-cell antibodies that cause demyelination122. Blood plasma, consisting of water, proteins, electrolytes, immunoglobulins, and other nutrients123, circulates in the body as a transport medium. However, other substances, such as abnormal antibodies, can also be present in the plasma, as in the case of Guillain-Barré Syndrome. TPE enables efficient and direct filtration of the plasma, removing up to 86% pathogenic antibodies124.
Though plasmapheresis is a well-established autoimmune disease treatment for neurological diseases, there have also been hypotheses and trials associating this treatment with neurodegenerative diseases such as Alzheimer’s, suggesting the potential of removing abnormal toxins and proteins that contribute to neurodegeneration125. Since Guillain-Barré syndrome is associated with the peripheral nervous system instead of the central nervous system, malignant antibodies are prevalent in the bloodstream as they do not need to cross the BBB during nervous system degradation. However, in Alzheimer’s, the BBB remains a major barrier between the bloodstream and the interstitium of the brain, impeding direct fluid and material exchanges across the membrane. Given this, scientists have targeted the potential application of TPE for Alzheimer’s disease based on the peripheral sink hypothesis, establishing that a dynamic equilibrium controls the amounts of Aβ protein in the cerebrospinal fluids and the brain126. Removal of Aβ protein from the bloodstream could cause the body to shift towards homeostatic balance, promoting the passive diffusion and active transport of Aβ from the central nervous system across the blood-brain barrier127. Furthermore, albumin, the most abundant protein found in the plasma128, could be used as a replacement protein during plasma exchange, for mobilisation and stabilisation of blood plasma, and to reduce pro-inflammatory cytokines129.
In a preclinical modelling trial utilising mouse brains to evaluate the applicability of the peripheral sink hypothesis, scientists performed albumin plasma exchange on mouse models to examine the effect on Aβ accumulation in the brain. After 7 months of TPE, researchers presented findings that reveal moderate benefits of applying TPE for removing Aβ in Alzheimer’s disease. Results showcased a significant decline in plasma Aβ, followed by a decrease in the quantity and size of insoluble protein plaques in the mouse brain upon biopsy analysis. Although no correlation was observed with soluble protein, the findings still support the proposition of cognitive benefits of TPE applied to Alzheimer’s disease130.
Nevertheless, given the variations between mouse brain models and human brains, deviations in efficacy and safety considerations may exist, and considerations of other factors, such as disease progression, existing medical conditions, and patient comfort, are needed. Applying this mechanism to a human clinical trial, Boada et al. conducted an 18-month study with 347 patients diagnosed with mild to moderate Alzheimer’s. Patients underwent albumin exchange and IV immunoglobulin, and were examined by both clinical and neuropsychological testing to determine physiopathological changes following the treatment. Results showed that within patients with mild Alzheimer’s, those who received TPE treatment showed higher ratings in numerous categories, such as daily life activity performance (ADAS-ADL) and cognitive subscale (ADAS-Cog)131. However, patients with moderate Alzheimer’s did not show an improvement in cognitive abilities when compared to the placebo group. Especially given the nature of Alzheimer’s disease to progress rapidly into the late stage, only 50.4% of all diagnosed patients have mild disease, suggesting a limited applicability of TPE for the entire population of Alzheimer’s patients132. Furthermore, 501 procedure-related adverse events were reported, including catheter reactions, hypotension, muscle spasm, and more, as well as psychological effects of anxiety. Two patients passed away during the period of the study, but whether or not the death was directly related to the treatment was not explicitly stated. These adverse effects are mainly due to the invasiveness of the procedure, as well as the nature of TPE requiring up to a 6-hour duration133, increasing risks of infection, hematoma, and hypotension134.
From the preclinical and clinical trials above, the potential potency of TPE applied to Alzheimer’s disease is clear, but more evidence and further development are needed to support the claim. Given that plasmapheresis is an invasive procedure, safety and patient well-being are still underlying issues, with an elevated risk profile of severe adversities, especially aggravated by the advanced ages of the target patient population. Lastly, though TPE showed significant signs of decreased Aβ in plasma, current methods are still unable to perform whole blood transfusions, due to risks of allergic reactions and infections135.
Gene therapy: Antisense Oligonucleotides
Antisense oligonucleotides (ASO) are synthetic single-stranded (SS) oligodeoxynucleotides commonly applied to genetic disorders that target the RNA transcription pathway to repress or inhibit protein synthesis. The compositions of ssDNA or RNA are chemically modified by sulfurization, altering the phosphodiester backbone to decrease the susceptibility of ASO to nuclease degradation. Oftentimes, ribose rings are further adapted by the addition of auxiliary substituents, increasing nuclease resistance and aiding the linkage process136. Further modifications of alternate backbones and targeting ligands can also be applied to customise ASO for varying targets and purposes (explored below)137.
Target therapy by ASO is applied to the treatment of Huntington’s disease, aimed at reducing mHtt protein synthesis. In the treatment for Huntington’s disease, chemically modified ASO is delivered via lumbar intrathecal injection, reaching the cerebrospinal fluid and circulating within the central nervous system. Upon entering the nucleus, the ASO recognises and binds to mHtt pre-mRNA by complementary base pairing, preventing further processing or modification of the pre-mRNA. ASO then recruits a ribonuclease H (RNase H), where the endonuclease enzymes break phosphodiester bonds in the mRNA backbone by hydrolysis, degrading the mRNA and preventing the progression of protein synthesis that produces mHtt. This process effectively regulates gene expression and directly prevents the production of toxic protein aggregates138.
With Alzheimer’s disease being heavily based upon abnormal protein production and aggregation, coupled with increasing evidence of genetic relevance, gene therapy and interventions may become cardinal in Alzheimer’s treatment. Beyond the Apo4E gene variant with high likelihood of predisposing Alzheimer’s disease due to disrupted neuroplasticity and potential links of A aggregation, a recent study by Nguyen et al. discovered another potential genetic cause of Alzheimer’s disease. By utilizing CRISPR technology, their team investigated intronic repeats and discovered that the CASP8-GGGAGAEXP allele was significantly associated with Alzheimer’s pathology, noting that the polyGR-encoding repeat expansion mutations lead to toxin protein aggregation139.
Given potential genetic relevance in Alzheimer’s, alongside the role of protein aggregation in its pathological pathway, a similar mechanism of ASO treatment could be applied, aimed at decreasing the production of A or tau proteins. An investigational ASO therapy targeting tau protein in Alzheimer’s, named BIIB080, was granted Fast Track designation by the FDA, and is currently in Phase II trial testing the efficacy and safety of this therapy140.
Similarly, another drug known as MAPTRx was also developed to target tau neurofibril by inhibiting the MAPT gene that produces tau proteins. Upon intrathecal injection, the ASO enters neuronal cells, binding to MAPT mRNA and recruiting RNaseH1 to carry out degradation. Similar to the mechanism of action for the HTT gene in Huntington’s disease, the ASO intervention inhibits protein synthesis, preventing tau protein formation. ASO is capable of multiple mechanisms, by inhibiting post-transcriptional modifications of mRNA, or promoting cleavage and destruction with RNaseH (Fig. 7).

In a 2023 phase I trial by Mummery et al., MAPTRx was tested with 34 patients with mild to moderate Alzheimer’s, with results concluding high efficacy of over 50% mean reduction from baseline tau protein concentration in the CSF, as well as a notable decrease of phosphorylated tau proteins142. Although further phase II and III trials are needed to confirm safety and efficacy at larger scales, it is clear that ASO has great potential to be implemented into Alzheimer’s treatment, directly targeting the pathophysiology of protein aggregation. However, the study also reported 94% of patients experiencing mild to moderate adverse effects, with the majority due to lumbar puncture, a crucial procedure for intrathecal drug delivery. Though not a severe adverse effect, this is still a significant limitation and drawback for ASO treatment due to its constant recurrence and inevitability.
Furthermore, ASO treatment poses the risk of systemic inflammatory responses, by potentially triggering immune responses by the synthetic nucleic acids and causing organ-specific toxicity and hematological abnormalities143. To mitigate this, numerous chemical modifications have been explored. Studies show that removing a carbonyl group to increase resistance to protease degradation, its neutral charge also helps avoid triggering an immune reaction144. Similarly, modifications such as 2′-O-methylation were shown to enhance molecular stability, while locked nucleic acid modification increased binding potency and durability145. Most recently, stereopure backbone linkages, including phosphorothioate (PS) and phosphoryl guanidine (PN) backbones, are arising as advantageous modifications, enhancing overall therapeutic potency and reducing toxicity146. At the same time, with recent advancements in ultrasound-assisted and nanoparticle-mediated delivery, incorporation could reduce the need for repeated spinal injections by enhancing the efficiency of each treatment delivery147. With the development and improvement of such technologies, the precision and efficiency of ASO treatment could be largely increased, while minimising adverse effects, thus enabling safer, more effective treatments for a wide range of disorders with a genetic basis.
Overview
Cross-over applications for Alzheimer’s disease, including Deep Brain Stimulation and Focused Ultrasound, Therapeutic plasma exchange, and gene therapy, are emerging therapeutic approaches, with increasing clinical trials proving their efficacy and benefits. Table 3 below summarises key clinical trials for each Alzheimer’s disease therapy discussed.
| Mechanism of action | Methodology | Trial outcomes* | Safety | |
| Deep Brain Stimulation | Implantation of electrodes to deliver electrical impulses that modulate dysfunctional neural circuits, potentially normalizing aberrant signaling and reducing symptoms. | Phase 1 trial with 6 Alzheimer’s patients with mild disease, receiving continuous DBS treatment for 12 months112 | – Evidence of increased neuronal activity (notable in entorhinal and hippocampal regions) – Non-conclusive changes to ADAS-cog scores (50% patients showing improvement and others worsening) – Increase in quality of life measure (4/6 patients) – Significant sustained increase in glucose metabolism in relevant brain regions | No serious adverse effect noted |
| Phase 1 trial with 20 Alzheimer’s patients with severe disease, subjected to postoperative stimulation and monitored for 12 months148 | – Significant improvement of MMSE scores noted – ADAS-cog score changes were not sustained long-term – NPI and HAMA scores indicate improved sleep quality and decreased neuropsychiatric symptoms | – 20% experienced mild adverse effects – 2 patients experienced severe subdural hematoma – 6 patients deceased during the follow-up period, but none with causes directly related to the trial | ||
| Therapeutic plasma exchange | Protein aggregates clearance from the bloodstream, reducing brain amyloid-beta (Aβ) levels by enhancing its efflux from the CNS. | Phase 2b/3 trial with 47 Alzheimer’s patients undergoing plasma exchange with albumin and intravenous immunoglobulin replacement130 | – Treated group showed a 52% less decline in daily function – Treatment group showed 66% less decline based on the ADAS-Cog scale – Biomarker analysis (Aβ42, T-tau, and P-tau) suggested that treatment prevented significant worsening of protein accumulation compared to placebo | – 10.6% of trials were associated with adverse effects – Serious adverse effects were reported to be more common in patients receiving intravenous immunoglobulin compared to albumin replacement |
| Antisense oligonucleotide gene therapy | ASO binding to the target RNA sequence inhibits protein expression by cleavage and degradation. | Phase 1b trial with 46 Alzheimer’s patients receiving varying amounts of MAPTRx drug (tau-targeting antisense oligonucleotide)142 | – 31% mean percentage decrease of t-tau concentration from baseline observed in the treatment group – Variations between treatment groups of different dosage amounts are evident – Notable sustained decline in CSF p-tau181 in two treatment groups | – 44% of patients reported adverse effects – 2 patients experienced mild stroke receiving a placebo |
*Abbreviations
ADAS-cog: Alzheimer’s Disease Assessment Scale-Cognitive Subscale
MMSE: Mini-Mental State Examination
NPI: Neuropsychiatric Inventory
HAMA: Hamilton Anxiety Scale
Discussion
This study has employed a comparative analysis of Parkinson’s, Huntington’s, and Guillain-Barré syndrome as a conceptual lens through which to re-examine the complex pathophysiology of Alzheimer’s disease. Close analysis reveals provocative parallels with Alzheimer’s, particularly in the domains of bidirectionality between the peripheral nervous system and the central nervous system, astrocytic dysfunction with neuroinflammation, and genetic predisposition. Subsequently, the explored approaches of deep-brain stimulation, therapeutic plasma exchange, and antisense oligonucleotide gene therapy have shown great potential in treatment applications for Alzheimer’s disease, with current clinical trials attesting to their efficacy.
It is, however, crucial to acknowledge that these parallels are not equivalences. The discussed diseases are vastly distinct in disease pathway, symptoms, and causes, and the proposed overlaps in this study do not concretely establish absolute similitude. Thus, existing fundamental differences may limit the long-term efficacy of extrapolative therapies for Alzheimer’s disease.
A proposed future direction is combination therapy, to design holistic treatment courses that maximise and integrate disparate yet complementary medicines and technologies149. This can be done by formulating multiphasic treatments that target distinct pathological pathways of the disease, achieving holistic therapeutic effects. Concurrently, precision medicine, also known as personalised medicine, has emerged as a novel approach to tailor therapies to individual patients150. With expanded possibilities of treatment approaches, precision medicine could be integrated to target patients’ idiosyncratic disease severities and pathological pathways.
Conclusion
Neurodegenerative disorders remain one of the most complex and perplexing fields of study in medicine, with elusive causes and severe limitations in investigation due to the complexity of the human brain and its interconnectedness with other critical mechanisms.
However, rapid clinical advancements in recent years have proven the high possibility of extraordinary achievements. As of 2025, a new therapy for Huntington’s disease demonstrated high efficacy and potential for eradicating the disorder. Similarly, as presented in this study, innovation through integrating perspectives and approaches could yield viable prospects and novel solutions. This study emphasises the complexity of this progressive neurodegenerative condition, demonstrating the necessity of further efforts in integrating multifaceted treatment options.
Still, despite the promise of novel treatments in clinical trials discussed in this study, challenges persist in the treatment of Alzheimer’s disease, and future research to corroborate and refine such approaches is needed. At the same time, this study focuses primarily on Guillain-Barré syndrome, Huntington’s disease, Parkinson’s Disease, and their relevance to Alzheimer’s disease, though the analysed pathological similarities may not have encompassed the full nuances of these diseases. Beyond concretising and investigating further relatedness between these diseases, future expansions could include exploring other neurological or neurodegenerative diseases and their current treatments, conducting applicative trials, and testing to evaluate implementation potential.
Furthermore, with the dynamic and ever-changing landscape of the scientific and medical field, versatility and receptivity will be crucial in order to capitalise on these advances, like integrating artificial intelligence in early diagnosis screening or therapy development. Ultimately, a deliberate commitment to inventive methodologies and the exploration of cross-modal integration is essential to generate novel, synergistic solutions that the field demands.
References
- World Health Organization. Dementia. World Health Organization https://www.who.int/news-room/fact-sheets/detail/dementia 2023 [↩]
- L. P. Gwyther. Social issues of the alzheimer’s patient and family. The American Journal of Medicine. Vol. 104, pg. 17S-21S, 1998 https://doi.org/10.1016/s0002-9343(98)00024-2 [↩]
- J. Zhang, Y. Zhang, J. Wang, Y. Xia, J. Zhang, L. Chen. Recent advances in alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduction and Targeted Therapy. Vol. 9, 2024 https://doi.org/10.1038/s41392-024-01911-3 [↩]
- Alzheimer’s Association. Stages of alzheimer’s. Alzheimer’s Disease and Dementia https://www.alz.org/alzheimers-dementia/stages 2024 [↩]
- National Institute on Aging. How is alzheimer’s disease treated? National Institute on Aging https://www.nia.nih.gov/health/alzheimers-treatment/how-alzheimers-disease-treated 2023 [↩]
- What happens to the brain in alzheimer’s disease? National Institute on Aging https://www.nia.nih.gov/health/alzheimers-causes-and-risk-factors/what-happens-brain-alzheimers-disease 2024 [↩]
- N. P. Dantuma, L. C. Bott. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Frontiers in Molecular Neuroscience. Vol. 7, 2014 https://doi.org/10.3389/fnmol.2014.00070 [↩]
- Y. Hsieh, Z. M. Augur, M. Arbery, N. Ashour, K. Barrett, R. V. Pearse, E. S. Tio, D. M. Duong, D. Felsky, P. L. De, D. A. Bennett, N. T. Seyfried, T. L. Young‐Pearse. Person‐specific differences in ubiquitin‐proteasome mediated proteostasis in human neurons. Alzheimer’s & Dementia. Vol. 20, 2024 https://doi.org/10.1002/alz.13680 [↩]
- A. M. Gorman. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. Journal of Cellular and Molecular Medicine. Vol. 12, pg. 2263–2280, 2008 https://doi.org/10.1111/j.1582-4934.2008.00402.x [↩]
- Y. Sun, H. Yu, Y. Guan. Glia connect inflammation and neurodegeneration in multiple sclerosis. Neuroscience Bulletin. Vol. 39, pg. 466–478, 2023 https://doi.org/10.1007/s12264-023-01034-9 [↩]
- N. J. Rothwell. Cytokines ‐ killers in the brain? The Journal of Physiology. Vol. 514, pg. 3–17, 1999 https://doi.org/10.1111/j.1469-7793.1999.003af.x [↩]
- J. Zhang, G. Kong, J. Yang, L. Pang, X. Li. Pathological mechanisms and treatment progression of alzheimer’s disease. European Journal of Medical Research. Vol. 30, 2025 https://doi.org/10.1186/s40001-025-02886-9 [↩]
- J. Singh, V. Raja, M. Irfan, O. Hashmat, M. Syed, N. N. Shahbaz. Frequency of autonomic dysfunction in patients of guillain barre syndrome in a tertiary care hospital. Cureus. Vol. 12, 2020 https://doi.org/10.7759/cureus.12101 [↩]
- World Health Organization. Guillain–barré syndrome. Who.int https://www.who.int/news-room/fact-sheets/detail/guillain-barr%C3%A9-syndrome 2023 [↩]
- S. M. Callahan, C. G. Dolislager, J. G. Johnson. The host cellular immune response to infection by campylobacter spp. and its role in disease. Infection and Immunity. Vol. 89, 2021 https://doi.org/10.1128/iai.00116-21 [↩]
- F. Bolukbasi, G. Ersen, A. Gunduz, F. Karaali Savrun, S. Yazici, N. Uzun, M. A. Akalin, M. E. Kiziltan. Guillain-barre syndrome and its variants: clinical course and prognostic factors. Noro Psikiyatri Arsivi. 2017 https://doi.org/10.5152/npa.2017.18091 [↩]
- R. Bellanti, S. Rinaldi. Guillain-barré syndrome: a comprehensive review. European Journal of Neurology. Vol. 31, 2024 https://doi.org/10.1111/ene.16365 [↩]
- H. Wekerle, H. Lassmann. The immunology of inflammatory demyelinating disease. McAlpine’s Multiple Sclerosis. pg. 491–555, 2006 https://doi.org/10.1016/B978-0-443-07271-0.50013-6 [↩]
- R. K. Yu, S. Usuki. Ganglioside molecular mimicry and its pathological roles in guillain-barré syndrome and related diseases. ASM Journals https://journals.asm.org/doi/10.1128/iai.00967-06 2006 [↩]
- A. R. Ferguson, J. R. Huie, E. D. Crown, K. M. Baumbauer, M. A. Hook, S. M. Garraway, K. H. Lee, K. C. Hoy, J. W. Grau. Maladaptive spinal plasticity opposes spinal learning and recovery in spinal cord injury. Frontiers in Physiology. Vol. 3, pg. 399, 2012 https://doi.org/10.3389/fphys.2012.00399 [↩]
- Z. Chen, Y. L. Balachandran, W. P. Chong, K. W. Y. Chan. Roles of cytokines in alzheimer’s disease. International Journal of Molecular Sciences. Vol. 25, pg. 5803, 2024 https://doi.org/10.3390/ijms25115803 [↩]
- M. E. Lull, M. L. Block. Microglial activation and chronic neurodegeneration. Neurotherapeutics. Vol. 7, pg. 354–365, 2010 https://doi.org/10.1016/j.nurt.2010.05.014 [↩]
- P. Vanacker, G. Matias, P. Hagmann, P. Michel. Cerebral hypoperfusion in posterior reversible encephalopathy syndrome is different from transient ischemic attack on ct perfusion. Journal of Neuroimaging : Official Journal of the American Society of Neuroimaging. Vol. 25, pg. 643–6, 2015 https://doi.org/10.1111/jon.12158 [↩]
- G. Nehra, B. Bauer, A. M. S. Hartz. Blood-brain barrier leakage in alzheimer’s disease: from discovery to clinical relevance. Pharmacology & Therapeutics. Vol. 234, pg. 108119, 2022 https://doi.org/10.1016/j.pharmthera.2022.108119 [↩]
- NIH. Huntington’s disease. National Institute of Neurological Disorders and Stroke https://www.ninds.nih.gov/health-information/disorders/huntingtons-disease 2022 [↩]
- H. Tong, T. Yang, S. Xu, X. Li, L. Liu, G. Zhou, S. Yang, S. Yin, X.-J. Li, S. Li. Huntington’s disease: complex pathogenesis and therapeutic strategies. International Journal of Molecular Sciences. Vol. 25, pg. 3845–3845, 2024 https://doi.org/10.3390/ijms25073845 [↩]
- G. Bitetto, A. Di Fonzo. Nucleo–cytoplasmic transport defects and protein aggregates in neurodegeneration. Translational Neurodegeneration. Vol. 9, 2020 https://doi.org/10.1186/s40035-020-00205-2 [↩]
- Szu Yi Chou, Ju Yun Weng, Hsing Lin Lai, F. Liao, S. H. Sun, Pang Hsien Tu, D. W. Dickson, Y. Chern. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (ccl5/rantes) by astrocytes. The journal of Neuroscience/The Journal of Neuroscience. Vol. 28, pg. 3277–3290, 2008 https://doi.org/10.1523/jneurosci.0116-08.2008 [↩]
- R. Wang, P. H. Reddy. Role of glutamate and nmda receptors in alzheimer’s disease. Journal of Alzheimer’s Disease. Vol. 57, pg. 1041–1048, 2017 https://doi.org/10.3233/jad-160763 [↩]
- N. Puranik, M. Song. Glutamate: molecular mechanisms and signaling pathway in alzheimer’s disease, a potential therapeutic target. Molecules. Vol. 29, pg. 5744–5744, 2024 https://doi.org/10.3390/molecules29235744 [↩]
- L.-L. Cao, P.-P. Guan, Y.-Y. Liang, X.-S. Huang, P. Wang. Calcium ions stimulate the hyperphosphorylation of tau by activating microsomal prostaglandin e synthase 1. Frontiers in Aging Neuroscience. Vol. 11, 2019 https://doi.org/10.3389/fnagi.2019.00108 [↩]
- E. J. Slow, R. K. Graham, M. R. Hayden. To be or not to be toxic: aggregations in huntington and alzheimer disease. Science Direct https://www.sciencedirect.com/science/article/abs/pii/S0168952506001685 2006 [↩]
- T. A. Thibaudeau, R. T. Anderson, D. M. Smith. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nature Communications. Vol. 9, 2018 https://doi.org/10.1038/s41467-018-03509-0 [↩]
- Z. Zhang, X. Yang, Y.-Q. Song, J. Tu. Autophagy in alzheimer’s disease pathogenesis: therapeutic potential and future perspectives. Ageing Research Reviews. Vol. 72, pg. 101464, 2021 https://doi.org/10.1016/j.arr.2021.101464 [↩]
- P. L. Brattås, B. A. Hersbach, S. Madsen, R. Petri, J. Jakobsson, K. Pircs. Impact of differential and time-dependent autophagy activation on therapeutic efficacy in a model of huntington disease. Autophagy. pg. 1–14, 2020 https://doi.org/10.1080/15548627.2020.1760014 [↩]
- E. Cho, M. Park. Palmitoylation in alzheimer⿿s disease and other neurodegenerative diseases. Pharmacological Research. Vol. 111, pg. 133–151, 2016 https://doi.org/10.1016/j.phrs.2016.06.008 [↩]
- K. Zhang, M. Wen, X. Nan, S. Zhao, H. Li, Y. Ai, H. Zhu. NMDA receptors in neurodegenerative diseases: mechanisms and emerging therapeutic strategies. Frontiers in Aging Neuroscience. Vol. 17, 2025 https://doi.org/10.3389/fnagi.2025.1604378 [↩]
- Tadahiro Numakawa, Ryutaro Kajihara. The role of brain-derived neurotrophic factor as an essential mediator in neuronal functions and the therapeutic potential of its mimetics for neuroprotection in neurologic and psychiatric disorders. Molecules. Vol. 30, pg. 848–848, 2025 https://doi.org/10.3390/molecules30040848 [↩]
- T. Alqahtani, S. L. Deore, A. A. Kide, B. A. Shende, R. Sharma, R. Dadarao Chakole, L. S. Nemade, N. Kishor Kale, S. Borah, S. Shrikant Deokar, A. Behera, D. Dhawal Bhandari, N. Gaikwad, A. Kalam Azad, A. Ghosh. Mitochondrial dysfunction and oxidative stress in alzheimer’s disease, and parkinson’s Disease, huntington’s disease and amyotrophic lateral sclerosis -an updated review. Mitochondrion. Vol. 71, pg. 83–92, 2023 https://doi.org/10.1016/j.mito.2023.05.007 [↩]
- M. Fathi, K. Vakili, Shirin Yaghoobpoor, Arian Tavasol, Kimia Jazi, Ramtin Hajibeygi, S. Shool, Fatemeh Sodeifian, Andis Klegeris, A. McElhinney, Mostafa Rezaei Tavirani, Fatemeh Sayehmiri. Dynamic changes in metabolites of the kynurenine pathway in alzheimer’s disease, parkinson’s Disease, and huntington’s disease: a systematic review and meta-analysis. Frontiers in Immunology. Vol. 13, 2022 https://doi.org/10.3389/fimmu.2022.997240 [↩]
- D. E. Ehrnhoefer, B. K. Y. Wong, M. R. Hayden. Convergent pathogenic pathways in alzheimer’s and huntington’s diseases: shared targets for drug development. Nature Reviews Drug Discovery. Vol. 10, pg. 853–867, 2011 https://doi.org/10.1038/nrd3556 [↩]
- D. Aarsland, M. W. Kurz. The epidemiology of dementia associated with parkinson’s Disease. Brain Pathology. Vol. 20, pg. 633–639, 2010 https://doi.org/10.1111/j.1750-3639.2009.00369.x [↩]
- A. S. Andrew, F. L. Anderson, S. L. Lee, K. M. Von Herrmann, M. C. Havrda. Lifestyle factors and parkinson’s Disease risk in a rural new england case-control study. Parkinson’s Disease. Vol. 2021, pg. 1–7, 2021 https://doi.org/10.1155/2021/5541760 [↩]
- M. T. Periñán, K. Brolin, S. Bandres-Ciga, C. Blauwendraat, C. Klein, Z. Gan-Or, A. Singleton, P. Gomez-Garre, M. Swanberg, P. Mir, A. Noyce. Effect modification between genes and environment and parkinson’s Disease risk. Annals of Neurology. Vol. 92, pg. 715–724, 2022 https://doi.org/10.1002/ana.26467 [↩]
- G. Yu, Q. Su, Y. Chen, L. Wu, S. Wu, H. Li. Epigenetics in neurodegenerative disorders induced by pesticides. Genes and Environment. Vol. 43, 2021 https://doi.org/10.1186/s41021-021-00224-z [↩]
- J. Tran, H. Anastacio, C. Bardy. Genetic predispositions of parkinson’s Disease revealed in patient-derived brain cells. Npj Parkinson’s Disease. Vol. 6, pg. 1–18, 2020 https://doi.org/10.1038/s41531-020-0110-8 [↩]
- K.-H. Chang, G.-J. Lee-Chen, Y.-R. Wu, Y.-J. Chen, J.-L. Lin, M. Li, I.-C. Chen, Y.-S. Lo, H.-C. Wu, C.-M. Chen. Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic parkinson’s Disease. Parkinsonism & Related Disorders. Vol. 24, pg. 81–8, 2016 https://doi.org/10.1016/j.parkreldis.2016.01.001 [↩]
- Z. Zhou, L. Yi, Q. Wang, T. M. Lim, E. K. Tan. Role of dopamine in the pathophysiology of parkinson’s Disease. Translational Neurodegeneration. Vol. 12, 2023 https://doi.org/10.1186/s40035-023-00378-6 [↩]
- M. Spano. The possible involvement of mitochondrial dysfunctions in lewy body dementia: a systematic review. Functional Neurology. 2015 https://doi.org/10.11138/fneur/2015.30.3.151 [↩]
- S. A. Tanik, C. E. Schultheiss, L. A. Volpicelli-Daley, K. R. Brunden, V. M. Y. Lee. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. Journal of Biological Chemistry. Vol. 288, pg. 15194–15210, 2013 https://doi.org/10.1074/jbc.m113.457408 [↩]
- C. B. Young, J. Sonne, V. Reddy. Neuroanatomy, basal ganglia. Nih.gov https://www.ncbi.nlm.nih.gov/books/NBK537141/ 2023 [↩]
- N. de O. Manzanza, L. Sedlackova, R. N. Kalaria. Alpha-synuclein post-translational modifications: implications for pathogenesis of lewy body disorders. Frontiers in Aging Neuroscience. Vol. 13, 2021 https://doi.org/10.3389/fnagi.2021.690293 [↩]
- V. Walecha, P. M. Luthra. The mitochondrial-astrocyte-neuron triad hypothesis in parkinson’s Disease: a toxic feedback loop of metabolism, aggregation, and oxidative stress. Neurochemical Research. Vol. 50, pg. 317, 2025 https://doi.org/10.1007/s11064-025-04559-9 [↩]
- W. Won, M. Bhalla, J.-H. Lee, C. J. Lee. Astrocytes as key regulators of neural signaling in health and disease. Annual Review of Neuroscience. Vol. 48, pg. 251–276, 2025 https://doi.org/10.1146/annurev-neuro-112723-035356 [↩]
- M. Huang, A. Long, L. Hao, Z. Shi, M. Zhang. Astrocyte in neurological disease: pathogenesis and therapy. MedComm. Vol. 6, 2025 https://doi.org/10.1002/mco2.70299 [↩]
- Y. Zhao, Y. Huang, Y. Cao, J. Yang. Astrocyte-mediated neuroinflammation in neurological conditions. Biomolecules. Vol. 14, pg. 1204–1204, 2024 https://doi.org/10.3390/biom14101204 [↩]
- Vicente Javier Clemente-Suárez, L. Redondo-Flórez, Ana Isabel Beltrán-Velasco, Domingo Jesús Ramos-Campo, P. Belinchón-deMiguel, I. Martínez-Guardado, A. A. Dalamitros, R. Yáñez-Sepúlveda, A. Martín-Rodríguez, José Francisco Tornero-Aguilera. Mitochondria and brain disease: a comprehensive review of pathological mechanisms and therapeutic opportunities. Biomedicines. Vol. 11, pg. 2488–2488, 2023 https://doi.org/10.3390/biomedicines11092488 [↩]
- C. Wang, T. Yang, M. Liang, J. Xie, N. Song. Astrocyte dysfunction in parkinson’s Disease: from the perspectives of transmitted α-synuclein and genetic modulation. Translational Neurodegeneration. Vol. 10, 2021 https://doi.org/10.1186/s40035-021-00265-y [↩]
- P.-P. Liu, Y. Xie, X.-Y. Meng, J.-S. Kang. History and progress of hypotheses and clinical trials for alzheimer’s disease. Signal Transduction and Targeted Therapy. Vol. 4, 2019 https://doi.org/10.1038/s41392-019-0063-8 [↩]
- J. Fortea, J. Pegueroles, D. Alcolea, O. Belbin, O. Dols-Icardo, L. Vaqué-Alcázar, L. Videla, J. D. Gispert, M. Suárez-Calvet, S. C. Johnson, R. Sperling, A. Bejanin, A. Lleó, V. Montal. APOE4 homozygozity represents a distinct genetic form of alzheimer’s disease. Nature Medicine. Vol. 30, pg. 1–8, 2024 https://doi.org/10.1038/s41591-024-02931-w [↩]
- Y. Li, J. R. Macyczko, C.-C. Liu, G. Bu. ApoE4 reduction: an emerging and promising therapeutic strategy for alzheimer’s disease. Neurobiology of Aging. 2022 https://doi.org/10.1016/j.neurobiolaging.2022.03.011 [↩]
- A. Jones, M. U. Ali, A. Mayhew, K. Aryal, R. H. Correia, D. Dash, D. R. Manis, A. Rehman, M. E. O’Connell, V. Taler, A. P. Costa, D. B. Hogan, C. Wolfson, P. Raina, L. Griffith. Environmental risk factors for all-cause dementia, alzheimer’s disease dementia, vascular dementia, and mild cognitive impairment: an umbrella review and meta-analysis. Environmental Research. Vol. 270, pg. 121007, 2025 https://doi.org/10.1016/j.envres.2025.121007 [↩]
- R. J. O’Brien, P. C. Wong. Amyloid precursor protein processing and alzheimer’s disease. Annual Review of Neuroscience. Vol. 34, pg. 185–204, 2011 https://doi.org/10.1146/annurev-neuro-061010-113613 [↩]
- S. Soriano, D. C. Lu, S. Chandra, C. U. Pietrzik, E. H. Koo. The amyloidogenic pathway of amyloid precursor protein (app) is independent of its cleavage by caspases. Journal of Biological Chemistry. Vol. 276, pg. 29045–29050, 2001 https://doi.org/10.1074/jbc.m102456200 [↩]
- A. Habib, D. Sawmiller, J. Tan. Restoring soluble amyloid precursor protein αβ functions as a potential treatment for alzheimer’s disease. Journal of Neuroscience Research. Vol. 95, pg. 973–991, 2016 https://doi.org/10.1002/jnr.23823 [↩]
- S. L. Cole, R. Vassar. The alzheimer’s disease beta-secretase enzyme, bace1. Molecular Neurodegeneration. Vol. 2, pg. 22, 2007 https://doi.org/10.1186/1750-1326-2-22 [↩]
- O. Engmann. Crosstalk between cdk5 and gsk3β: implications for alzheimer’s disease. Frontiers in Molecular Neuroscience. Vol. 2, 2009 https://doi.org/10.3389/neuro.02.002.2009 [↩]
- G. Paterno, B. M. Bell, K.-M. M. Gorion. Reassessment of neuronal tau distribution in adult human brain and implications for tau pathobiology. BMC https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-022-01394-9#:~:text=Our%20findings%20demonstrate%20that%20under,is%20predominantly%20comprised%20of%20axons 2022 [↩]
- A. Mietelska-Porowska, U. Wasik, M. Goras, A. Filipek, G. Niewiadomska. Tau protein modifications and interactions: their role in function and dysfunction. International Journal of Molecular Sciences. Vol. 15, pg. 4671–4713, 2014 https://doi.org/10.3390/ijms15034671 [↩]
- C. A. Lasagna-Reeves, D. L. Castillo-Carranza, U. Sengupta, A. L. Clos, G. R. Jackson, R. Kayed. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Molecular Neurodegeneration. Vol. 6, pg. 39, 2011 https://doi.org/10.1186/1750-1326-6-39 [↩]
- C. Marcus, E. Mena, R. M. Subramaniam. Brain pet in the diagnosis of alzheimer’s disease. Clinical Nuclear Medicine. Vol. 39, pg. e413–e426, 2014 https://doi.org/10.1097/rlu.0000000000000547 [↩]
- T. Pflanzner, M. C. Janko, B. André-Dohmen, S. Reuss, S. Weggen, A. J. M. Roebroek, C. R. W. Kuhlmann, C. U. Pietrzik. LRP1 mediates bidirectional transcytosis of amyloid-β across the blood-brain barrier. Neurobiology of Aging. Vol. 32, pg. 2323.e1-2323.e11, 2011 https://doi.org/10.1016/j.neurobiolaging.2010.05.025 [↩]
- A. S. Haqqani, K. Bélanger, D. B. Stanimirovic. Receptor-mediated transcytosis for brain delivery of therapeutics: receptor classes and criteria. Frontiers in Drug Delivery. Vol. 4, 2024 https://doi.org/10.3389/fddev.2024.1360302 [↩]
- D. Yetman. Can a pet scan diagnose alzheimer’s disease? Healthline https://www.healthline.com/health/alzheimers/pet-scan-for-alzheimers 2023 [↩]
- Moeko Noguchi‐Shinohara, K. Shuta, H. Murakami, Y. Mori, J. Komatsu, C. Kobayashi, S. Hersch, K. Horie, K. Ono. Lecanemab‐Associated amyloid‐β protofibril in cerebrospinal fluid correlates with biomarkers of neurodegeneration in alzheimer’s disease. Annals of Neurology. 2025 https://doi.org/10.1002/ana.27175 [↩]
- R. Tarawneh, G. D’Angelo, E. Macy, C. Xiong, D. Carter, N. J. Cairns, A. M. Fagan, D. Head, M. A. Mintun, J. H. Ladenson, J.-M. Lee, J. C. Morris, D. M. Holtzman. Visinin-like protein-1: diagnostic and prognostic biomarker in alzheimer disease. Annals of Neurology. Vol. 70, pg. 274–285, 2011 https://doi.org/10.1002/ana.22448 [↩]
- G. Grande, M. Valletta, D. Rizzuto, X. Xia, C. Qiu, N. Orsini, M. Dale, S. Andersson, C. Fredolini, B. Winblad, E. J. Laukka, L. Fratiglioni, D. L. Vetrano. Blood-based biomarkers of alzheimer’s disease and incident dementia in the community. Nature Medicine. 2025 https://doi.org/10.1038/s41591-025-03605-x [↩]
- Adjanie Patabendige, Damir Janigro. The role of the blood–brain barrier during neurological disease and infection. Biochemical Society Transactions. Vol. 51, 2023 https://doi.org/10.1042/bst20220830 [↩]
- J. Sun, Z. He. Interactions between astrocytes and cerebral endothelial cells in central nervous system diseases. Biomedicine & Pharmacotherapy. Vol. 191, pg. 118516, 2025 https://doi.org/10.1016/j.biopha.2025.118516 [↩]
- L. A. Khawli, S. Prabhu. Drug delivery across the blood–brain barrier. Molecular Pharmaceutics. Vol. 10, pg. 1471–1472, 2013 https://doi.org/10.1021/mp400170b [↩]
- R. M. Shah, S. R. Jadhav, G. Bryant, I. P. Kaur, I. H. Harding. On the formation and stability mechanisms of diverse lipid-based nanostructures for drug delivery. Advances in Colloid and Interface Science. Vol. 338, pg. 103402, 2025 https://doi.org/10.1016/j.cis.2025.103402 [↩]
- X. Cai, C. J. Drummond, J. Zhai, N. Tran. Lipid nanoparticles: versatile drug delivery vehicles for traversing the blood brain barrier to treat brain cancer. Advanced Functional Materials. 2024 https://doi.org/10.1002/adfm.202404234 [↩]
- M. Gionso, E. Herlin, L. Uva, F. Guidi, P. Tortoli, G. Durando, L. Raspagliesi, N. Corradino, V. Percuoco, F. DiMeco, M. De Curtis, L. Librizzi, F. Prada. Ultrasound guided blood brain barrier opening using a diagnostic probe in a whole brain model. Scientific Reports. Vol. 15, 2025 https://doi.org/10.1038/s41598-025-94660-4 [↩]
- Y.-S. Tung, F. Vlachos, J. A. Feshitan, M. A. Borden, E. E. Konofagou. The mechanism of interaction between focused ultrasound and microbubbles in blood-brain barrier opening in mice. The Journal of the Acoustical Society of America. Vol. 130, pg. 3059–3067, 2011 https://doi.org/10.1121/1.3646905 [↩]
- J. M. Wasielewska, A. R. White. “Focused ultrasound-mediated drug delivery in humans – a path towards translation in neurodegenerative diseases”. Pharmaceutical Research. Vol. 39, pg. 427–439, 2022 https://doi.org/10.1007/s11095-022-03185-2 [↩]
- J. Chen, P. Xiang, A. Duro-Castano, H. Cai, B. Guo, X. Liu, Y. Yu, S. Lui, K. Luo, B. Ke, L. Ruiz-Pérez, Q. Gong, X. Tian, G. Battaglia. Rapid amyloid-β clearance and cognitive recovery through multivalent modulation of blood–brain barrier transport. Signal Transduction and Targeted Therapy. Vol. 10, pg. 1–12, 2025 https://doi.org/10.1038/s41392-025-02426-1 [↩]
- J. H. Pedder, A. M. Sonabend, M. D. Cearns, B. D. Michael, R. Zakaria, A. B. Heimberger, M. D. Jenkinson, D. Dickens. Crossing the blood–brain barrier: emerging therapeutic strategies for neurological disease. The Lancet Neurology. Vol. 24, 2025 https://doi.org/10.1016/s1474-4422(24)00476-9 [↩]
- S. Chowdhury, Nurjahan Shipa Chowdhury. Novel anti-amyloid-beta (aβ) monoclonal antibody lecanemab for alzheimer’s disease: a systematic review. International Journal of Immunopathology and Pharmacology. Vol. 37, 2023 https://doi.org/10.1177/03946320231209839 [↩]
- B.-H. Kim, S. Kim, Y. Nam, Y. H. Park, S. M. Shin, M. Moon. Second-generation anti-amyloid monoclonal antibodies for alzheimer’s disease: current landscape and future perspectives. Translational Neurodegeneration. Vol. 14, 2025 https://doi.org/10.1186/s40035-025-00465-w [↩]
- D. A. Loeffler. Antibody-mediated clearance of brain amyloid-β: mechanisms of action, effects of natural and monoclonal anti-aβ antibodies, and downstream effects. Journal of Alzheimer’s Disease Reports. Vol. 7, pg. 873–899, 2023 https://doi.org/10.3233/adr-230025 [↩]
- C. H. van Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer, T. Iwatsubo. Lecanemab in early alzheimer’s disease. New England Journal of Medicine. Vol. 388, pg. 9–21, 2022 https://doi.org/10.1056/nejmoa2212948 [↩]
- N. J. Reish, P. Jamshidi, B. Stamm, M. E. Flanagan, E. Sugg, M. Tang, K. L. Donohue, M. McCord, C. Krumpelman, M.-M. Mesulam, R. Castellani, S. H.-Y. Chou. Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t-pa for stroke. New England Journal of Medicine. 2023 https://doi.org/10.1056/nejmc2215148 [↩]
- J. L. Cummings, A. M. Leisgang, D. Cammann, J. Powell, J. Chen. Anti-amyloid monoclonal antibodies for the treatment of alzheimer’s disease. BioDrugs. Vol. 38, 2023 https://doi.org/10.1007/s40259-023-00633-2 [↩]
- R. S. Doody, R. Raman, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, F. He, X. Sun, R. G. Thomas, P. S. Aisen, E. Siemers, G. Sethuraman, R. Mohs. A phase 3 trial of semagacestat for treatment of alzheimer’s disease. New England Journal of Medicine. Vol. 369, pg. 341–350, 2013 https://doi.org/10.1056/nejmoa1210951 [↩]
- L. Guha, N. Singh, H. Kumar. Challenges in drug development for neurological disorders. Springer Nature Link. pg. 27–45, 2023 https://doi.org/10.1007/978-981-99-6807-7_2 [↩]
- C. J. Swanson, Y. Zhang, S. Dhadda, J. Wang, J. Kaplow, R. Y. K. Lai, L. Lannfelt, H. Bradley, M. Rabe, A. Koyama, L. Reyderman, D. A. Berry, S. Berry, R. Gordon, L. D. Kramer, J. L. Cummings. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early alzheimer’s disease with lecanemab, an anti-aβ protofibril antibody. Alzheimer’s Research & Therapy. Vol. 13, 2021 https://doi.org/10.1186/s13195-021-00813-8 [↩]
- J. Cummings,
L. Apostolova, G. D. Rabinovici, A.
Atri, P. Aisen, S. Greenberg, S. Hendrix,
D. Selkoe, M. Weiner, R. C. Petersen,
S. Salloway. Lecanemab: appropriate
use recommendations. The Journal of
Prevention of Alzheimer’s Disease.
Vol. 10, 2023 https://doi.org/10.14283
/jpad.2023.30 [↩] - S. Salloway, A. Pain, E. Lee, M. Papka, M. B. Ferguson, H. Wang, H. Hu, M. Lu, E. Oru, P. A. Ardayfio, D. B. Hoban, E. C. Collins, D. A. Brooks, J. R. Sims. TRAILBLAZER‐ALZ 4: a phase 3 trial comparing donanemab with aducanumab on amyloid plaque clearance in early, symptomatic alzheimer’s disease. Alzheimer s & Dementia. Vol. 21, 2025 https://doi.org/10.1002/alz.70293 [↩]
- G. D. Rabinovici, D. J. Selkoe, S. E.
Schindler, P. Aisen, L. G. Apostolova,
A. Atri, S. M. Greenberg, S. B. Hendrix,
R. C. Petersen, M. Weiner, S. Salloway,
J. Cummings. Donanemab: appropriate
use recommendations. The Journal of
Prevention of Alzheimer’s Disease. Vol.
12, pg. 100150, 2025 https://doi.org/
10.1016/j.tjpad.2025.100150 [↩] - M. Meglio. Eli lilly’s remternetug demonstrates significant amyloid plaque removal in early-stage trial. Neurology live https://www.neurologylive.com/view/eli-lilly-remternetug-demonstrates-significant-amyloid-plaque-removal-early-stage-trial 2023 [↩]
- ALZ forum: roche spells out phase three plans for trontinemab. Globalalzplatform.org https://globalalzplatform.org/2025/08/22/alz-forum-roche-spells-out-phase-three-plans-for-trontinemab/ 2025 [↩]
- Hans Peter Grimm, V. Schumacher, M. Schäfer, S. Imhof-Jung, Per‐Ola Freskgård, K. Brady, C. Hofmann, P. Rüger, T. Schlothauer, Ulrich Göpfert, M. J. Hartl, S. Rottach, A. Zwick, S. Seger, R. Neff, J. Niewoehner, N. Janssen. Delivery of the brainshuttleTM amyloid-beta antibody fusion trontinemab to non-human primate brain and projected efficacious dose regimens in humans. mAbs. Vol. 15, 2023 https://doi.org/10.1080/19420862.2023.2261509 [↩]
- J. Theuriet, C. Aguesse, F. Bouhour, L. Jomir, S. Thobois, S. Prange. Guillain-barré syndrome following subthalamic nucleus – deep brain stimulation in parkinson’s Disease: a case report. Revue Neurologique. Vol. 180, pg. 459–461, 2023 https://doi.org/10.1016/j.neurol.2023.09.002 [↩]
- L. Wojtecki, S. Groiss, C. Hartmann, S. Elben, S. Omlor, A. Schnitzler, J. Vesper. Deep brain stimulation in huntington’s disease—preliminary evidence on pathophysiology, efficacy and safety. Brain Sciences. Vol. 6, pg. 38, 2016 https://doi.org/10.3390/brainsci6030038 [↩]
- Can Sarica, C. R. Conner, K. Yamamoto, A. Yang, J. Germann, M. Lannon, N. Samuel, M. Colditz, B. Santyr, C. Chow, C. Iorio-Morin, D. H. Aguirre-Padilla, S. Lang, Artur Vetkas, C. Cheyuo, A. Loh, Ghazaleh Darmani, O. Flouty, V. Milano, M. Paff, Mojgan Hodaie, S. K. Kalia, R. P. Munhoz, A. Fasano, A. M. Lozano. Trends and disparities in deep brain stimulation utilization in the united states: a nationwide inpatient sample analysis from 1993 to 2017. The Lancet Regional Health – Americas. Vol. 26, pg. 100599–100599, 2023 https://doi.org/10.1016/j.lana.2023.100599 [↩]
- Y. Smith. What does deep brain stimulation involve? News-Medical.net https://www.news-medical.net/health/What-does-deep-brain-stimulation-involve.aspx 2015 [↩]
- S. Chiken, A. Nambu. Mechanism of deep brain stimulation. The Neuroscientist. Vol. 22, pg. 313–322, 2015 https://doi.org/10.1177/1073858415581986 [↩]
- A. L. Benabid. Deep brain stimulation for parkinson’s Disease. Current Opinion in Neurobiology. Vol. 13, pg. 696–706, 2003 https://doi.org/10.1016/j.conb.2003.11.001 [↩]
- H. J. MacDonald, W. D. Byblow. Does response inhibition have pre- and postdiagnostic utility in parkinson’s Disease? Journal of Motor Behavior. Vol. 47, pg. 29–45, 2015 https://doi.org/10.1080/00222895.2014.941784 [↩]
- B. Picton, J. Wong, A. M. Lopez, S. S. Solomon, S. Andalib, N. J. Brown, R. R. Dutta, M. R. Paff, F. P. Hsu, M. Y. Oh. Deep brain stimulation as an emerging therapy for cognitive decline in alzheimer’s disease: systematic review of evidence and current targets. World Neurosurgery. Vol. 184, pg. S1878-8750(23)01817, 2023 https://doi.org/10.1016/j.wneu.2023.12.083 [↩]
- C. Hamani, M. P. McAndrews, M. Cohn, M. Oh, D. Zumsteg, C. M. Shapiro, R. A. Wennberg, A. M. Lozano. Memory enhancement induced by hypothalamic/fornix deep brain stimulation. Annals of Neurology. Vol. 63, pg. 119–123, 2008 https://doi.org/10.1002/ana.21295 [↩]
- A. W. Laxton, D. F. Tang-Wai, M. P. McAndrews, D. Zumsteg, R. Wennberg, R. Keren, J. Wherrett, G. Naglie, C. Hamani, G. S. Smith, A. M. Lozano. A phase i trial of deep brain stimulation of memory circuits in alzheimer’s disease. Annals of Neurology. Vol. 68, pg. 521–534, 2010 https://doi.org/10.1002/ana.22089 [↩] [↩]
- I. Arrieta-Cruz, C. Pavlides, G. Pasinetti. Deep brain stimulation in midline thalamic region facilitates synaptic transmission and short-term memory in a mouse model of alzheimer’s disease. Translational Neuroscience. Vol. 1, 2010 https://doi.org/10.2478/v10134-010-0023-x [↩]
- M. Olson, H. A. Shill, F. A. Ponce, S. Aslam. Deep brain stimulation in pd: risk of complications, morbidity, and hospitalizations: a systematic review. Frontiers in Aging Neuroscience. Vol. 15, 2023 https://doi.org/10.3389/fnagi.2023.1258190 [↩]
- A. Kim, H.-J. Yang, J.-H. Kwon, M.-H. Kim, J. Lee, B. Jeon. Mortality of deep brain stimulation and risk factors in patients with parkinson’s Disease: a national cohort study in korea. Journal of Korean Medical Science. Vol. 38, pg. e10, 2023 https://doi.org/10.3346/jkms.2023.38.e10 [↩]
- L. Morgante, F. Morgante, E. Moro, A. Epifanio, P. Girlanda, P. Ragonese, A. Antonini, P. Barone, U. Bonuccelli, M. F. Contarino, L. Capus, M. G. Ceravolo, R. Marconi, R. Ceravolo, M. D’Amelio, G. Savettieri. How many parkinsonian patients are suitable candidates for deep brain stimulation of subthalamic nucleus? results of a questionnaire. Parkinsonism & Related Disorders. Vol. 13, pg. 528–531, 2007 https://doi.org/10.1016/j.parkreldis.2006.12.013 [↩]
- Y. Luo, Y. Sun, X. Tian, X. Zheng, X. Wang, W. Li, X. Wu, B. Shu, W. Hou. Deep brain stimulation for alzheimer’s disease: stimulation parameters and potential mechanisms of action. Frontiers in Aging Neuroscience. Vol. 13, 2021 https://doi.org/10.3389/fnagi.2021.619543 [↩]
- X. Liu, S. S. M. Naomi, W. L. Sharon, E. J. Russell. The applications of focused ultrasound (fus) in alzheimer’s disease treatment: a systematic review on both animal and human studies. Aging and Disease. Vol. 12, pg. 1977–2002, 2021 https://doi.org/10.14336/AD.2021.0510 [↩]
- S.-H. Liu, Yi Long Lai, Bo Lin Chen, F. Yang. Ultrasound enhances the expression of brain-derived neurotrophic factor in astrocyte through activation of trkb-akt and calcium-camk signaling pathways. Cerebral Cortex. Vol. 27, pg. bhw169–bhw169, 2016 https://doi.org/10.1093/cercor/bhw169 [↩]
- R. Beisteiner, E. Matt, C. Fan, H. Baldysiak, M. Schönfeld, T. Philippi Novak, A. Amini, T. Aslan, R. Reinecke, J. Lehrner, A. Weber, U. Reime, C. Goldenstedt, E. Marlinghaus, M. Hallett, H. Lohse‐Busch. Transcranial pulse stimulation with ultrasound in alzheimer’s disease—a new navigated focal brain therapy. Advanced Science. Vol. 7, pg. 1902583, 2019 https://doi.org/10.1002/advs.201902583 [↩]
- K. Eguchi, T. Shindo, K. Ito, T. Ogata, R. Kurosawa, Y. Kagaya, Y. Monma, S. Ichijo, S. Kasukabe, S. Miyata, T. Yoshikawa, K. Yanai, H. Taki, H. Kanai, N. Osumi, H. Shimokawa. Whole-brain low-intensity pulsed ultrasound therapy markedly improves cognitive dysfunctions in mouse models of dementia – crucial roles of endothelial nitric oxide synthase. Brain Stimulation. Vol. 11, pg. 959–973, 2018 https://doi.org/10.1016/j.brs.2018.05.012 [↩]
- A. Stoian, G. Șerban, Z. Bajko, S. Andone, O. Mosora, A. Bălașa. Therapeutic plasma exchange as a first‑choice therapy for axonal guillain‑barré syndrome: a case‑based review of the literature (review). Experimental and Therapeutic Medicine. Vol. 21, 2021 https://doi.org/10.3892/etm.2021.9696 [↩]
- J. Mathew, M. Varacallo, P. Sankar. Physiology, blood plasma. Nih.gov https://www.ncbi.nlm.nih.gov/books/NBK531504/ 2023 [↩]
- F. Foettinger, G. Pilz, P. Wipfler, A. Harrer, J. M. Kern, E. Trinka, T. Moser. Immunomodulatory aspects of therapeutic plasma exchange in neurological disorders—a pilot study. International Journal of Molecular Sciences. Vol. 24, pg. 6552–6552, 2023 https://doi.org/10.3390/ijms24076552 [↩]
- J. I. Spencer, M. Crane, M. Pisa, A. D. Waldman, G. C. DeLuca. Out with the old, in with the new: could plasma exchange be used to fill a therapeutic gap in neurology? Journal of the Neurological Sciences. Vol. 432, pg. 120056–120056, 2021 https://doi.org/10.1016/j.jns.2021.120056 [↩]
- Y. Yi, J. Lee, M. H. Lim. Amyloid-β-interacting proteins in peripheral fluids of alzheimer’s disease. Trends in Chemistry. Vol. 6, pg. 128–143, 2024 https://doi.org/10.1016/j.trechm.2024.01.003 [↩]
- B. P. Imbimbo, S. Ippati, F. Ceravolo, M. Watling. Perspective: is therapeutic plasma exchange a viable option for treating alzheimer’s disease? Alzheimer’s & Dementia: Translational Research & Clinical Interventions. Vol. 6, 2020 https://doi.org/10.1002/trc2.12004 [↩]
- R. N. Moman, M. Varacallo, N. Gupta. Physiology, albumin. Nih.gov https://www.ncbi.nlm.nih.gov/books/NBK459198/ 2022 [↩]
- R. Gonzalo, C. Minguet, A. M. Ortiz, M. I. Bravo, O. L. López, M. Boada, A. Ruiz, M. Costa. Plasma exchange with albumin replacement for alzheimer’s disease treatment induced changes in serum and cerebrospinal fluid inflammatory mediator levels. Annals of Clinical and Translational Neurology. 2024 https://doi.org/10.1002/acn3.52235 [↩]
- S. Ramirez, S. Koerich, N. Astudillo, N. De Gregorio, R. Al-Lahham, T. Allison, N. P. Rocha, F. Wang, C. Soto. Plasma exchange reduces aβ levels in plasma and decreases amyloid plaques in the brain in a mouse model of alzheimer’s disease. International Journal of Molecular Sciences. Vol. 24, pg. 17087, 2023 https://doi.org/10.3390/ijms242317087 [↩] [↩]
- M. Boada, O. L. López, J. Olazarán, L. Núñez, M. Pfeffer, M. Paricio, J. Lorites, G. Piñol‐Ripoll, J. E. Gámez, F. Anaya, D. Kiprov, J. Lima, C. Grifols, M. Torres, M. Costa, J. Bozzo, Z. M. Szczepiorkowski, S. Hendrix, A. Páez. A randomized, controlled clinical trial of plasma exchange with albumin replacement for alzheimer’s disease: primary results of the ambar study. Alzheimer’s & Dementia. Vol. 16, pg. 1412–1425, 2020 https://doi.org/10.1002/alz.12137 [↩]
- J. Yuan, N. Maserejian, Y. Liu, S. Devine, C. Gillis, J. Massaro, R. Au. Severity distribution of alzheimer’s disease dementia and mild cognitive impairment in the framingham heart study. Journal of Alzheimer’s Disease. Vol. 79, pg. 807–817, 2021 https://doi.org/10.3233/jad-200786 [↩]
- I. Baldwin, S. Todd. Therapeutic plasma exchange in the intensive care unit and with the critically ill, a focus on clinical nursing considerations. Journal of Clinical Apheresis. Vol. 37, pg. 397–404, 2022 https://doi.org/10.1002/jca.21984 [↩]
- S. Baruah, S. Periyavan. Adverse effects of intermittent flow therapeutic plasmapheresis in neurology patients in a resource constrained setting. Global Journal of Transfusion Medicine. Vol. 4, pg. 163, 2019 https://doi.org/10.4103/gjtm.gjtm_1_19 [↩]
- C. Elena Cervantes, E. M. Bloch, C. John Sperati. Therapeutic plasma exchange: core curriculum 2023. American Journal of Kidney Diseases. Vol. 81, pg. 475–492, 2023 https://doi.org/10.1053/j.ajkd.2022.10.017 [↩]
- Ruchi Ruchi, G. M. Raman, V. Kumar, Raman Bahal. Evolution of antisense oligonucleotides: navigating nucleic acid chemistry and delivery challenges. Expert Opinion on Drug Discovery. 2024 https://doi.org/10.1080/17460441.2024.2440095 [↩]
- S. T. Crooke, T. A. Vickers, X. Liang. Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acids Research. Vol. 48, pg. 5235–5253, 2020 https://doi.org/10.1093/nar/gkaa299 [↩]
- A. V. Smith, S. J. Tabrizi. Therapeutic antisense targeting of huntingtin. DNA and Cell Biology. Vol. 39, pg. 154–158, 2020 https://doi.org/10.1089/dna.2019.5188 [↩]
- L. Nguyen, R. Ajredini, S. Guo, L. E. L. Romano, R. F. Tomas, L. R. Bell, P. T. Ranum, T. Zu, M. Bañez Coronel, C. P. Kelley, J. Redding-Ochoa, E. Nizamis, A. Melloni, T. R. Connors, A. Gaona, K. Thangaraju, O. Pletnikova, H. B. Clark, B. L. Davidson, A. T. Yachnis, T. E. Golde, X. Lou, E. T. Wang, A. E. Renton, A. Goate, P. N. Valdmanis, S. Prokop, J. C. Troncoso, B. T. Hyman, L. P. W. Ranum. CASP8 intronic expansion identified by poly-glycine-arginine pathology increases alzheimer’s disease risk. Proceedings of the National Academy of Sciences. Vol. 122, 2025 https://doi.org/10.1073/pnas.2416885122 [↩]
- Biogen’s investigational tau-targeting therapy biib080 receives fda fast track designation for the treatment of alzheimer’s disease | biogen. Biogen https://investors.biogen.com/news-releases/news-release-details/biogens-investigational-tau-targeting-therapy-biib080-receives 2025 [↩]
- A. L. Southwell, N. H. Skotte, C. F. Bennett, M. R. Hayden. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends in Molecular Medicine. Vol. 18, pg. 634–643, 2012 https://doi.org/10.1016/j.molmed.2012.09.001 [↩]
- C. J. Mummery, A. Börjesson-Hanson, D. J. Blackburn, E. G. B. Vijverberg, P. P. De Deyn, S. Ducharme, M. Jonsson, A. Schneider, J. O. Rinne, A. C. Ludolph, R. Bodenschatz, H. Kordasiewicz, E. E. Swayze, B. Fitzsimmons, L. Mignon, K. M. Moore, C. Yun, T. Baumann, D. Li, D. A. Norris, R. Crean, D. L. Graham, E. Huang, E. Ratti, C. F. Bennett, C. Junge, R. M. Lane. Tau-targeting antisense oligonucleotide maptrx in mild alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nature Medicine. Vol. 29, pg. 1–11, 2023 https://doi.org/10.1038/s41591-023-02326-3 [↩] [↩]
- K. S. Frazier. Antisense oligonucleotide therapies. Toxicologic Pathology. Vol. 43, pg. 78–89, 2014 https://doi.org/10.1177/0192623314551840 [↩]
- J. Lee, T. Yokota. Antisense therapy in neurology. Journal of Personalized Medicine. Vol. 3, pg. 144–176, 2013 https://doi.org/10.3390/jpm3030144 [↩]
- M. Herkt, T. Thum. Pharmacokinetics and proceedings in clinical application of nucleic acid therapeutics. Molecular Therapy. Vol. 29, pg. 521–539, 2021 https://doi.org/10.1016/j.ymthe.2020.11.008 [↩]
- K. Chwalenia, M. J. A. Wood, T. C. Roberts. Progress and prospects in antisense oligonucleotide-mediated exon skipping therapies for duchenne muscular dystrophy. Journal of Muscle Research and Cell Motility. pg. 10.1007/s10974-024, 2025 https://doi.org/10.1007/s10974-024-09688-2 [↩]
- E. Perolina, S. Meissner, B. Raos, B. Harland, S. Thakur, D. Svirskis. Translating ultrasound-mediated drug delivery technologies for cns applications. Advanced Drug Delivery Reviews. Vol. 208, pg. 115274–115274, 2024 https://doi.org/10.1016/j.addr.2024.115274 [↩]
- J. Xu, B. Liu, G. Shang, Z. Feng, H. Yang, Y. Chen, X. Yu, Z. Mao. Efficacy and safety of bilateral deep brain stimulation (dbs) for severe alzheimer’s disease: a comparative analysis of fornix versus basal ganglia of meynert. CNS Neuroscience & Therapeutics. Vol. 31, pg. e70285, 2025 https://doi.org/10.1111/cns.70285 [↩]
- J. L. Cummings, A. H. Burstein, H. Fillit. Alzheimer combination therapies: overview and scenarios. The Journal of Prevention of Alzheimer’s Disease. Vol. 12, pg. 100328, 2025 https://doi.org/10.1016/j.tjpad.2025.100328 [↩]
- FDA. Precision medicine. FDA https://www.fda.gov/medical-devices/in-vitro-diagnostics/precision-medicine 2019 [↩]



{kind=link}